
Introduction
The process of integrating clinical trial data for an Integrated Summary of Safety (ISS) or an Integrated Summary of Efficacy (ISE) is a challenging undertaking for clinical trial sponsors. Several factors must be considered, including choosing the proper studies for inclusion, selecting an optimal integration strategy, addressing variations across coding dictionaries, and understanding different dosing schedules and visit windowing.1,2,3,4
Integration efforts can quickly become more daunting if real-world data (RWD), defined by the Food and Drug Administration (FDA) as “data relating to individual patient health status or the delivery of health care routinely collected from a variety of sources,” are introduced.5 The FDA lists examples of RWD as data from electronic health records, medical claims data, data from product and disease registries, patient-generated data (including data from in-home-use settings), and data from other sources that can inform on health status such as mobile devices.5
Specializing in the rare disease space, Ultragenyx has developed Disease Monitoring Programs (DMPs), which are a new model of registries designed to monitor long-term disease manifestations. Within DMPs, patients can be on a sponsored drug, on another treatment, or not treated at all. These longitudinal, ten-year programs are designed to be comprehensive, good-clinical-practice-monitored formats where all measurements, tests, patient travel, and physician efforts are covered by the sponsoring company to assure that data are collected consistently as scheduled.6
To support and facilitate faster downstream analytics, we undertook a program-level initiative to integrate various data sources from clinical trials and DMPs, following Clinical Data Interchange Standards Consortium (CDISC) standards. Traceability and assurance of CDISC conformance were prioritized as essential throughout the development and execution of the integration strategy.
Although publications from PHUSE,1 PharmaSUG,2,3 and a CDISC draft guidance for Analysis Data Model (ADaM) Structures for Integration4 support pooling data from various sources, there is no unified approach to date within the industry on how to best integrate datasets from a standards perspective. Through our integration effort of aggregating both Study Data Tabulation Model (SDTM) and ADaM level datasets, using both clinical trials and RWD, we share this case study to highlight where data standards were expressive and facilitative to our integration needs. We also discuss areas of ambiguity within data standards encountered during the integration effort.
The objective of this integration project was to create comprehensive and fully harmonized datasets for clinical trial and RWD that uphold CDISC standards. By seamlessly linking the totality of the data for every subject, the datasets were designed to maximize future analysis needs.
Materials and Methods
Scope and study inclusion
Our program-level integration effort comprised over a decade’s worth of data that spanned 13 studies. These included phases I–III, a retrospective chart review, and the addition of an ongoing, post-marketing DMP. Subjects were allowed to enroll in multiple studies, and the characteristics from each are summarized in Table 1.7,8,9,10,11,12,13,14,15,16,17,18 The DMP study is a long-term outcomes program for subjects on or off any treatment to prospectively investigate change over time in biomarkers, clinical assessments, and patient or caregiver-reported outcomes measures.18 Some, but not all, of the endpoints collected from the clinical trials may be collected in the DMP, and other new endpoints may be collected in the DMP that were not obtained from earlier trials.
Individual Study Characteristics of the Integrated Case Study.
| Phase | Population | Design | Control Arm | Study Duration |
|---|---|---|---|---|
| 1* | Adult7 | Double-Blind | Placebo | <2 months |
| Adult8 | Open-Label | Placebo | 2 months | |
| Adult9 | Open-Label | Placebo | 13.5 months | |
| Adult10 | Open-Label | NA | <2 months | |
| 2 | Pediatric11 | Open-Label | NA | 13 to 18 months |
| Adult12 | Open-Label | NA | 14 to 16 months | |
| Adult13 | Open-Label | NA | 20 months | |
| 3 | Pediatric14 | Open-Label | Active Control | 5 months |
| Pediatric15 | Open-Label | Active Control | 5 to 11 months | |
| Adult16 | Double-Blind | Placebo | 8 to 13 months | |
| Adult17 | Open-Label | NA | 8 months | |
| Other | Pediatric | Retrospective Chart Review | NA | NA |
| Pediatric and Adult18 | Prospective – Real-World Setting (DMP Study) | NA – subjects can elect to be on or off treatment (including approved treatment or standard of care) | 10 years |
-
* Phase 1 Studies were conducted by a different sponsor.
NA = Not Applicable.
Integration methodology and strategy
We explored various options for how best to structure and build the integration framework itself (summarized in Table 3). Because this project was not required as part of a marketing application, we initially considered circumventing the use of CDISC to be free of the rigor required for implementing full standards (option 1). However, we selected to follow and uphold industry standards as the appeal of having the freedom to develop an internal standard likely would have been short-lived and difficult to maintain over time, leading to possible analysis errors. Aware that future data managers, programmers, and statisticians would be well-versed with established CDISC standards, we realized that it would be advantageous to build our integration framework on these fundamentals. Our primary aim was to uphold the concept of traceability.20 Leveraging the industry-wide familiarization with data standards would optimize future warehousing and data mining activities.
We created an independent, fully integrated set of SDTM domains using the individual studies’ SDTM domains as input. This decision was strongly influenced by the complexity of this project. Incongruencies existed across the studies’ SDTM domains, including dissimilar data collection systems, different sponsors, fluctuating SDTM programming teams, and the evolution of CDISC guidelines over the period during which the studies were conducted. Our goals for the integrated SDTM data were to
align similar data within the most appropriate domain (regardless whether this was done consistently across previous studies);
harmonize domain-level groupings (e.g., categories, test names, test codes);
perform harmonizing transformations, such as unit conversions where necessary, and medical and drug coding with consistent terminology; and
develop a rubric to ensure traceability back to the source study. If there was ambiguity regarding how to definitively classify, categorize, or harmonize the data, we would make a choice based on facilitating integration from the ongoing DMP study.
These new SDTM datasets were then used as the data sources to build integrated ADaM datasets, where both the integrated SDTM datasets and integrated ADaM datasets adhered as closely as possible to CDISC standards. The data flow is illustrated in Figure 3. At the ADaM level, our goals were to
optimize dataset readability for statisticians and clinicians;
execute anticipated visit windowing scenarios;
perform imputations where applicable; and
proactively define expected baseline(s), population sub-groups, and other projected analysis-supporting indicator variables.
Results
Our integration effort resulted in the generation of 16 SDTM datasets and 22 ADaM datasets containing data for over 1,000 rare disease subjects across two different therapeutic areas. Seventeen percent (n = 170) of subjects participated in more than one integrated study (maximum of five studies), and 71% of subjects are currently enrolled in the ongoing DMP. Adverse events, medical histories, and concomitant medications collected as part of the closed studies were up-coded to the same dictionary versions as the DMP study. Multiple types of clinical outcome assessments (COA) as defined by the Food and Drug Administration (FDA) were included (Table 2).21 The number of variables across all SDTM domains ranged from 14 (within the Disposition domain) to 47 (within the Laboratory Test Results domain), with the largest domain (Morphology) containing over 270,000 records. Across all ADaM datasets, 1,824 variables were created, with ADSL containing the greatest number of variables in a single dataset (270). Our largest ADaM dataset, ADLBCHEM, contains laboratory findings with nearly 150,000 integrated chemistry panel test results.
Sources of Collected Data.
| Domain (Name) | Tests, Assessments, Or Results Included | Data Source | |
|---|---|---|---|
| Structured CRF (EDC) | External Data | ||
| AE (Adverse Events) | Adverse Event Details (e.g., Severity, Outcome, Casualty, Action(s) Taken, etc.), Standard Coding Dictionary | X | |
| BR (Biopsy) | Bone Biopsy (i.e., Location, Grade, Interpretation, etc.) | X | |
| CM (Concomitant and Prior Medications) | Concomitant† and Prior Medications, Standard Coding Dictionary | X | X |
| DM (Demographics) | Basic Demographic Information (Full or Partial Dates of Birth, Gender, Race, etc.) | X | |
| DS (Disposition) | Date of Study Completion, Reason For Discontinuation, Death Details |
X | |
| DX (Device Exposure) | Device Details, Device Changes* |
X | |
| EC (Exposure as Collected) | Dose, Route of Administration, Dose Adjustment, Included Comparators, Placebo and Active Drug | X | X |
| EG (ECG Test Results) | Electrocardiogram Tests | X | X |
| EX (Exposure) | Dose, Route of Administration, Dose Adjustment, Included Comparators, Placebo and Active Drug | X | X |
| FA (Findings About) | Family History, Fatigue Diary |
X | |
| FT (Functional Test) | Six-Minute Walk Test, Timed Up and Go, Hand-Held Dynamometry |
X | |
| HO (Healthcare Encounters) | Office Visits and Rehabilitation* | X | |
| IS (Immunogenicity Specimen Assessment) | Antibody Testing (Drug, HIV, Hepatitis) | X | |
| LB (Laboratory Test Results) | Chemistry, Hematology, Urinalysis, Pregnancy Tests, Biomarkers |
X | X |
| MH (Medical History) | Family History, General History, Disease-Specific History,* Skeletal History,* Fracture History, Surgical History, Dental History |
X | |
| ML (Meal Data) | Timing,† Types of Foods Consumed,† Vitamin Range Consumed† |
X | X |
| MO (Morphology) | Renal Ultrasound, X-rays, CT Scans, Echocardiogram, Imaging Scans |
X | X |
| PC (Pharmacokinetics Concentrations) | Concentration Level, Timing, and Methodology | X | X |
| PE (Physical Exam) | Physical Exam Findings, Neurological Exam Results |
X | |
| PP (Pharmacokinetics Parameters) | Derived Pharmacokinetic Parameters (e.g., Area Under the Curve, Cmax, Tmax). | X | |
| PR (Procedures) | Medical, Dental, and Surgical Procedures | X | |
| QS (Questionnaires) | Clinical Interview*: Satisfaction of treatment, Clinical Interview*: Disease-Specific Conditions of Childhood Work and School Status,* Retirement and Disability Status,* Brief Pain Index,† Brief Fatigue Inventory,† SF-36 Questionnaire, SF-10 Questionnaire, Western Ontario and McMaster Universities Osteoarthritis Index, Patient Global Impression of Improvement, Patient Global Impression of Severity, Pediatric Outcomes Data Collection Instrument, Patient-Reported Outcomes Measurement Information System (PROMIS) |
X | X |
| RP (Reproductive System Findings) | Menopausal Status, Tanner Staging, Pregnancy History* |
X | |
| SU (Substance Use) | Additional Therapy (Nutrition) | X | |
| VS (Vital Signs) | Temperature, Blood Pressure, Respiration Rate, Weight, etc. |
X | |
| Custom Domain – (Genetic Mutations) | Mutation Results | X | X |
-
† eDiary Data.
* DMP Exclusively Collected Data.
Various Integration Strategy Approaches.
| Option | Integration Framework | Pro | Con |
|---|---|---|---|
| 1 | Integrate without using CDISC standards | • Allows for more fluid and flexible data integration • Permits the possibility of having only one set of integrated datasets |
• Likely a steep learning curve downstream for future data managers and programmers • Difficult to control derivation rule/variable naming/documentation creep as the project matures without a foundation data standard construct |
| 2 | Create integrated SDTMs using the individual studies’ raw1 data as the source | • Upholds direct traceability from source data to SDTM • Enables implementation of updated CDISC standards compared to what was released at the time of the original study |
• An immense amount of duplicative work as each study already has its own body of SDTMs created |
| 3* | Create integrated SDTMs using the individual studies’ SDTM domains as the source | • Reduction in duplicative work • Allows for harmonization and consistencies to be present within an integrated set of SDTM domains for easier ADaM processing • Enables implementation of updated CDISC standards compared to what was released at the time of the original study |
• Internal tools (e.g., macros and metadata files) not developed to facilitate this set-up • CDISC SDTM origin metadata for variables not directly upheld • There is no CDISC published SDTM level integration document released for review yet |
| 4 | Create integrated ADaMs using the individual studies’ SDTM domains as the source | • Permits the possibility of having only one set of integrated datasets | • Requires harmonizing differences amongst SDTM domains at the ADaM level in addition to the project-specific ADaM needs • Lack of integrated source data results in traceability back to individual studies’ SDTM with inconsistent data mapping schemes |
| 5 | Create integrated ADaMs using the individual studies’ ADaM domains as the source | • Potential to reduce the necessity to rederive certain calculations | • Failure to capitalize on the harmonization efforts put forth within the integrated SDTM domains • If there were dissimilar derivation rules required for integration than used for original studies this would be difficult to derive |
| 6* | Create integrated ADaMs using the integrated SDTM domains as the source | • Upholds direct traceability from integrated SDTM to integrated ADaMs • Easier ADaM creation process with data already integrated into SDTM |
• A large number of variables and records to hold new derivations and ensure traceability |
| 7 | Create integrated ADaMs using the individual studies’ raw1 data as the source | • Easier issue identification process with only two layers of data • Avoid the SDTM harmonization challenges |
• Convoluted ADaM creation process involving both data harmonization and analysis derivation • Likely a steep learning curve downstream for future data managers and programmers |
-
1 Defined as source EDC and external vendor data transfers.
* Selected as the final methodology for the project.
Discussion
Traceability
Traceability was upheld within the creation of the integrated SDTMs from the individual study SDTMs by way of a customized metadata file. For each domain, this file listed whether the variable was altered during integration and, if so, the new derivation rule. Additionally, all source SDTM data were retained in the integrated SDTM domains, mostly by using the SUPPQUAL datasets. For example, a subject enrolled in more than one study would have multiple informed consent dates, first dose dates, last dose dates, ages, and treatment arms collected across the various trials and in RWD. We kept the originally collected values in the SUPPDM.RFICDTCx, SUPPDM.RFXSTDTx, SUPPDM.RFXENDTx, SUPPDM.AGEx, SUPPDM.ARMx variables, where x ranged from 1 to 5 representing the original data sources. While SUPPQUAL is not designed for this purpose, we found it the most straightforward way to accomplish traceability back to the source study. However, there is no mechanism to trace the parent domain variable back to its SUPPQUAL origin. In our case, we elected a global rule to map into the parent domain the earliest date of collection for each subject when there was repeated information collected over the various trials. So in the case of the informed consent, although all informed consent dates were stored in SUPPDM, the earliest date was represented in DM.RFICDTC. In a large integration effort like this, extra documentation, including global rules, decisions, mapping specifications, and detailed conversion rules, are necessary to uphold traceability.
Integration challenges
We encountered systemic data collection variations stemming from different phase-specific and study design methodologies. Depending on the primary objective(s) of the individual study, data collection for a certain area of interest may be broad or specific, and in our experience, data collection tends to mature and evolve to become more specific over the lifetime of a program. For example, medical history collected from the first-in-human study captured the date the medical history was obtained, a yes/no checkbox for assessment of 12 (general) body systems, and an open text box for relevant history details. In the Phase 2 study, the medical history data collection expanded to include relatedness to disease state, start date, duration, and a slightly different representation of body systems, as seen in Figure 1. For the DMP post-marketing commitment, the medical history data collection grew to include multiple distinct forms, including the open collection of general body systems, detailed disease-specific common conditions of childhood, adulthood comorbidities, specific skeletal history, and dental history, as seen in Figure 2.
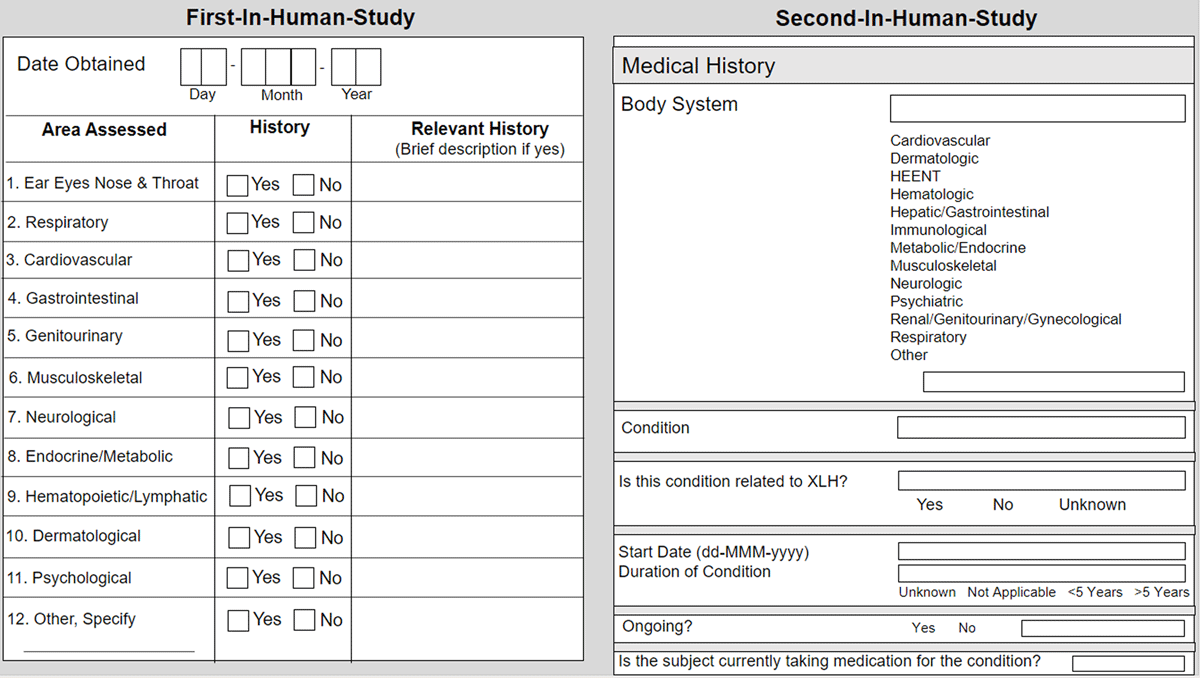
Examples of Medical History Case Report Forms for the First Two Clinical Studies.

Examples of Medical History Case Report Forms for the Post-Marketing Commitment (DMP Study).
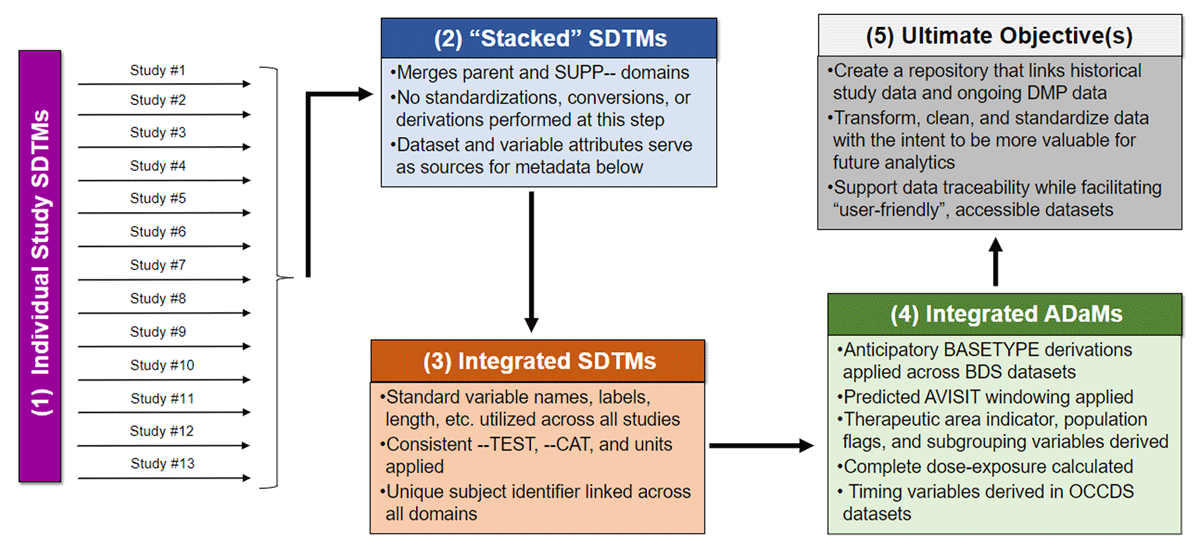
Flow Diagram Depicting the Construction of the Integrated Datasets.
Incorporating RWD and using it in an integrated approach to generate real-world evidence (RWE) warrants additional data harmonization considerations. RWE is defined by the FDA as “the clinical evidence regarding the usage and potential benefits or risks of a medical product derived from the analysis of RWD.”19 The DMP is a long-term study amongst both treated and non-treated subjects, which incorporates a visit schedule of either every six months or yearly that is determined by subject status (e.g., treated vs non-treated; adult vs pediatric). The discordant visit schedule translates into data collection and analysis challenges. As one example, at each bi-yearly or yearly visit a form is collected for every approved drug administration since the previous visit. Also, the DMP includes a mixture of both retrospective and prospective questions at a single visit. Thus, if a previous diagnosis of a certain condition was known, a series of questions related to worsening and planned surgeries and treatments are asked; however, if at the visit the diagnosis of the condition is not previously known, a different set of questions are raised. Due to the longevity of the study, there is a high likelihood of a single subject’s diagnosis status changing from unknown to known. Lastly, unlike in an ISS/ISE integration effort for submission to a regulatory body, our initiative aimed to harmonize all data elements (domains), and we operated without a clear analysis directive, which was especially challenging at the ADaM-level as there was no Statistical Analysis Plan (SAP).
CDISC standards that are supportive for integration
The use of CDISC Controlled Terminology (CT) was valuable during our pooling efforts. In our real-world application, as an example, body length measurements were collected using a variety of differing positions. Because these are clearly defined terms within the Position codelist (e.g., ‘DECUBITUS’, ‘STANDING’, SITTING’), the CT simplified the necessary transformations needed for data analysis, z-score, and percentile reporting.22
The Analysis Data Model Implementation Guide (ADaMIG) allows for multiple BASETYPEs within a domain. This allowance was paramount in our construction of ADaM datasets. As it is not uncommon to have more than one baseline definition in an integrated analysis of multiple treatments, BASETYPE enabled us to store multiple sets of records in a single domain. We want to highlight that allowing different derivations of the BASE, CHG, and PCHG for the same parameter (PARAM) in the BDS structure facilitates an intuitive way to filter and review the data by end-users. Without the allowance of multiple BASETYPEs, we would have considered either the creation of additional domains or the creation of additional variables (e.g. BASE1, BASE2, CHG1, CHG2, etc.) in the single domain, which defeats the purpose of the BDS structure. Both alternate solutions require the end-user to be fully proficient with the representation of the incrementation (e.g., ADVS1 vs ADVS2 or BASE1 vs BASE2 within ADVS).
Recommendations for future developments
Improved clarity of traceability from SDTM to analysis dataset construction
Although the Study Data Tabulation Model Implementation Guide (SDTMIG) definition for USUBJID is unambiguous and necessary in an integrated setting, the potential for issues when pooling subject-level data still exists.23 Specifically, within the Demographics (DM) domain, because subject-level values can change across studies, it is not clear how best to uphold traceability. We noticed that even conventionally static reported demographic variables can differ across studies (e.g., ETHNIC). In a large-scale integration effort where RWD may exist and where many clinical studies are complete, it is challenging or even impossible to understand the root cause of the perceived data contradiction. For example, was ETHNIC inadvertently recorded (and if so, which study?), or had the subject changed their self-reported ethnic identity over time? The potential presence of contradictory data as collected also exists beyond DM, particularly for historical recall where dates or dosages may not be accordant (e.g., prior medications and medical history forms). In such cases, we selected to integrate the initially reported value.
Because the variable –OBJ is unique to Findings About (FA),23 this domain was difficult to integrate. As a compounding factor, we discovered that FA was not always created consistently across studies, and even when the same data elements were mapped to FA, FAOBJ was not assigned reliably across studies. Although the SDTMIG contains a section to clarify when to use FA, in reality it appeared to be a slightly more subjective endeavor, whereby some programming teams upheld the spirit of the IG but others failed to appreciate the nuances of this domain. Instead, they (improperly) mapped FAOBJ data to supplemental qualifiers (SUPPFA), a custom domain, or the Medical History (MH) domain. The rheumatoid arthritis history example in Section 6.4 of the SDTMIG illustrates the vacillating qualities sometimes seen in this domain. In SDTMIG 3.2, the data are mapped to the FA domain.23 However, in SDTMIG 3.4, the guidance has been updated to map the same questions to the MH domain.24 In RWD settings, it is common to collect historical findings and events associated with the disease state, both at baseline and across evaluation intervals. Compiling this type of data for integration, including the amalgamation of previous mapping schemes and the recognition of updated guidance, introduced additional challenges in reestablishing harmonized data specifications. We selected to address these complications at the ADaM-level based on specific and sometimes evolving analysis needs.
The current ADaMIG for ADSL states that the dataset “contains one record per subject and does not fully cover the integration of multiple studies.”25 Between 2018–2019 there was a draft version of ADaM Structures for Integration released, but no further developments have been shared to our knowledge. Additionally, there has been nothing officially released for SDTM integration. A 2018 PharmaSUG paper details the proposal for new data structure classes for complex ADaM integration, including an integrated ADSL with multiple records per subject and corresponding integrated Occurrence Data Structure (OCCDS) and Base Data Structure (BDS) examples.26 Our integrated ADSL was constructed as one record per subject based upon overall pooling. It was not complicated to link from our integrated DM domain, as it was also structured as one record per unique subject. Arguably more important, we found that even for a complex integration, this structure was adaptable and manageable in practice. It supported clear reference for pivotal milestones (e.g., first treatment start date, each study start date) that enabled linkage to OCCDS or BDS data structures, but also permitted an intuitive yet comprehensible view of each subject’s data journey.
Consideration of controlled terminology from a regulatory perspective
For the variable RACE, the SDTMIG23,24 refers sponsors to FDA guidance and dictates that if multiple races are collected, then the value of RACE should be “MULTIPLE” with additional information included in the supplemental qualifiers dataset.27 The SDTMIG states that if race was collected via an “Other, specify” field, and the sponsor chooses not to map, then the value of race should be “OTHER.” However, the codelist for Race is non-extensible and does not support either “MULTIPLE” or “OTHER” as CDISC submission values. Although Race is a non-extensible codelist, we mapped the value of race in such cases as “OTHER” and supplied the additional race information to SUPPDM.
The definition of LBSTRESC is “Contains the result value for all findings, copied or derived from LBORRES in a standard format or standard units.”24 However, there is no clear directive as to what constitutes an expected standard unit from a regulator’s perspective. The current definition may work fine in a single study setting, but it is flawed when applied in an integrated setting whereby a sponsor, lab, or even protocol differences may contribute to different standard units being reported for the same lab test. For example, serum calcium may have mg/dL reported as the standard unit within one study but mmol/L in another, and both comply with the CDISC submission values for the Unit codelist. We elected the assistance of a medical monitor to determine the standard units for the specific disease state in which to report for our analysis needs.
Relaxing variable length limitations
The conformance limitation to maintain a variable name length to no more than eight characters is too restrictive in an integrated setting. The restriction stems from the requirement to submit SAS Transport Format Version 5 files28 and should be reconsidered for, at a minimum, an ISS/ISE data package. For example, within ADSL we needed to create variables that represent the 1,25-Dihydroxyvitamin D value at each of the original protocol-defined baselines and derive a new baseline defined as the last measure before first exposure to active drug. We aimed to combine the LBTESTCD (i.e., VITDAT), the two-digit padded integer to represent the period (i.e., xx), and the fragment to represent the baseline (i.e., BL). We were forced to drop the “BL” fragment in the variable name for each period (e.g., VITDATBL, VITDAT01, VITDAT02…). For clarity, the denotation of baseline for the period representing variables was included as part of their label descriptions. However, when assembling the same type of variables for shorter LBTESTCDs (e.g., PTHI), we were able to uphold the necessary data elements across all similar baseline variables within eight characters (e.g., PTHIBL, PTHIBL01, PTHIBL02…).
Current RWD Guidance
The FDA’s draft guidance for data standards involving RWD acknowledges the potential use of RWD to support the approval of new indications and satisfy post-approval requirements.5 Registries and DMPs have long been expected to monitor long-term, real-world data. Additionally, clinical trials have become more invested in wearable devices and hand-held devices for subject direct entry to contribute to the submission and include subject experience. The guidance suggests having each sponsor document the decisions made in mapping, but there is a need for pre-emptive variable name and dataset mapping guidance. Subjects may be asked to recall and enter recent fracture-related events into an eDiary. The collection may range from fracture occurrence (potential CE domain), pain scale data (potential QS domain), and other findings related to the fracture event (potential FA domain). The eDiary likely collects data entry date/time, completion date/time, delivery date/time, and fracture occurrence date/time. As these data likely will be mapped to different SDTM general observations classes for regulatory submission purposes, it is a challenge to try to retrofit eDiary data/time entries when pooling the data (i.e., –STDTC/–ENDTC are not present in findings domains). As for integration purposes, it may be more beneficial to keep the fracture-related diary data together in a cohesive domain rather than having it continue to be spread across many domains. More detailed and specific SDTM variable naming and domain mapping guidance would not only improve consistent reporting, but also sponsors would benefit also by saving time with submission preparations if ambiguity were removed. General rules could be applied to varying devices and study-specific collections from build to submission datasets.
From a data management perspective, wearable devices also present a challenge with the volume of data collected and stored, especially if the devices are worn continuously. For example, with Continuous Glucose Monitoring, values are collected every five minutes, resulting in over 100,000 records per subject, per year. Depending on the number of subjects and duration of the use of the device within the study, this necessitates storage and file management strategies, including data transfers and multiple files for the same data that exceeds storage limits and the additional time to use and manage these files. This data is considered SDTM (raw) data, which requires that all data collected for the same variables used in the analysis would be presented in the listings. Ideally, if there could be a reduction in what was submitted and presented to only the key timepoints or parameters used for analysis, with a complete, easily navigated, electronic version available for reference, this could assist with the final package creation and allow for a more manageable presentation of the data with less noise and submission burden.
Conclusion
From our experience, upholding CDSIC standards while integrating fully harmonized datasets from clinical trial data and RWD, we found that guidance and implementation guides are beginning to address the complexities of RWD. However, there is a need for further elucidation in some areas.
Additional direction for data handling surrounding multiple study enrollments by a single subject is needed. This includes how much (duplicated) data should be retained and documented, including the situations where the same data elements may not share the same reported result. A connection should be established between an integrated DM and an integrated ADSL, including how that relationship retains a connection when either one or both datasets are structured for either one or multiple records for a single subject.
Acknowledging that sponsors may dictate what is a standard unit for reporting, a preference for what regulators expect for standard laboratory findings would be facilitative for integrated submission packages. Direction is needed in the presentation of the voluminous amount of data received from the continuous collection of wearable devices and hand-held devices that are becoming the norm of studies to gather the subject’s full experience.
For sponsors, the creation of an ISS/ISE data package prepared for regulatory approval is just the first stage of a larger and often more complex integration effort that supports further scientific enrichment. The ideal for comprehensive end-to-end data standards should build upon defined regulatory requirements for integrated datasets that extend seamlessly to facilitate full program-level integration.
Acknowledgements
The authors would like to gratefully acknowledge Ultragenyx senior leadership for their support throughout this project.
Competing Interests
JS and KL are employees and shareholders of Ultragenyx Pharmaceutical Inc. DS is a consultant for Ultragenyx Pharmaceutical Inc.
References
1. U.S. Food and Drug Administration. Integrated Summary of Effectiveness Guidance for Industry. https://www.fda.gov/media/72335/download. Accessed May 3, 2022.
2. PharmaSUG. Best Practices for ISS/ISE Dataset Development. https://www.pharmasug.org/proceedings/2019/AD/PharmaSUG-2019-AD-299.pdf. Accessed May 3, 2022.
3. Lex Jansen. Navigating FDA requirement: ISS/ISE build strategies. https://www.lexjansen.com/phuse/2019/sa/SA07.pdf. Accessed May 3, 2022.
4. CDISC. ADaM Structures for Integration (CDISC Draft Guidance). https://wiki.cdisc.org/download/attachments/82586549/Meeting_2019-04-25_ADaM_Integration_and_Upcoming_Releases_Deborah%20Bauer.pdf?version=1&modificationDate=1561567396371&api=v2. Accessed May 3, 2022.
5. U.S. Food and Drug Administration. Data Standards for Drug and Biological Product Submissions Containing Real-World Data Guidance for Industry – Draft Guidance. https://www.fda.gov/media/153341/download. Accessed January 5, 2022.
6. Lochmüller H, Ramirez AN, Kakkis E. Disease monitoring programs of rare genetic diseases: transparent data sharing between academic and commercial stakeholders. Orphanet J Rare Dis. 2021 Mar 20; 16(1): 141. DOI: http://doi.org/10.1186/s13023-021-01687-7
7. NIH. A Study of KRN23 in X-linked Hypophosphatemia. https://clinicaltrials.gov/ct2/show/NCT00830674?cond=xlh&draw=3&rank=20. Accessed May 3, 2022.
8. NIH. A Repeated Study of KRN23 in Adults With X-Linked Hypophosphatemia. https://clinicaltrials.gov/ct2/show/NCT01340482?recrs=e&cond=xlh&phase=0&draw=2&rank=5. Accessed May 3, 2022.
9. NIH. An Extension Study of KRN23 in Adults With X-Linked Hypophosphatemia. https://clinicaltrials.gov/ct2/show/NCT01571596?recrs=e&cond=xlh&phase=0&draw=2&rank=4. Accessed May 3, 2022.
10. NIH. A Study of KRN23 in Subjects With X-linked Hypophosphatemic Rickets/Osteomalacia. https://clinicaltrials.gov/ct2/show/NCT02181764?recrs=e&cond=xlh&phase=0&draw=2&rank=2. Accessed May 3, 2022.
11. NIH. Study of KRN23 (Burosumab), a Recombinant Fully Human Monoclonal Antibody Against Fibroblast Growth Factor 23 (FGF23), in Pediatric Subjects With X-linked Hypophosphatemia (XLH). https://clinicaltrials.gov/ct2/show/NCT02163577?recrs=e&cond=xlh&age=0&phase=1&draw=2&rank=1. Accessed May 3, 2022.
12. NIH. Long-Term Extension Study of KRN23 in Adult Subjects With X-Linked Hypophosphatemia (XLH). https://clinicaltrials.gov/ct2/show/NCT02312687?recrs=e&cond=xlh&phase=1&draw=2&rank=1. Accessed May 3, 2022.
13. NIH. Study of Burosumab (KRN23) in Adults With Tumor-Induced Osteomalacia (TIO) or Epidermal Nevus Syndrome (ENS). https://clinicaltrials.gov/ct2/show/NCT02304367?recrs=e&cond=TIO&age=1&phase=1&draw=2&rank=1. Accessed May 3, 2022.
14. NIH. A Study of KRN23 in Pediatric Patients With X-linked Hypophosphatemic Rickets/Osteomalacia. https://clinicaltrials.gov/ct2/show/NCT03233126?recrs=e&cond=XLH&age=0&phase=2&fund=2&draw=2&rank=3. Accessed May 3, 2022.
15. NIH. Efficacy and Safety of Burosumab (KRN23) Versus Oral Phosphate and Active Vitamin D Treatment in Pediatric Patients With X Linked Hypophosphatemia (XLH). https://clinicaltrials.gov/ct2/show/NCT02915705?recrs=e&cond=XLH&age=012&phase=2&draw=2&rank=3. Accessed May 3, 2022.
16. NIH. Study of KRN23 in Adults With X-linked Hypophosphatemia (XLH). https://clinicaltrials.gov/ct2/show/NCT02526160?recrs=e&cond=XLH&age=012&phase=2&draw=2&rank=2. Accessed May 3, 2022.
17. NIH. Open Label Study of KRN23 on Osteomalacia in Adults With X-linked Hypophosphatemia (XLH). https://clinicaltrials.gov/ct2/show/NCT02537431?recrs=e&cond=XLH&age=012&phase=2&draw=2&rank=1. Accessed May 3, 2022.
18. NIH. X-linked Hypophosphatemia Disease Monitoring Program. https://clinicaltrials.gov/ct2/show/NCT03651505?cond=XLH&fund=2&draw=2&rank=8.
19. U.S. Food and Drug Administration. Submitting Documents Using Real-World Data and Real-World Evidence to FDA for Drugs and Biologics Guidance for Industry – Draft Guidance. https://www.fda.gov/media/124795/download. Accessed January 5, 2022.
20. CDISC. CDISC Analysis Data Model Implementation Guide Version 1.2. https://www.cdisc.org/standards/foundational/adam/adamig-v12. Accessed January 5, 2022.
21. U.S. Food and Drug Administration. Clinical Outcome Assessment (COA): Frequently Asked Questions. https://www.fda.gov/about-fda/clinical-outcome-assessment-coa-frequently-asked-questions#COADefinition. Accessed January 5, 2022.
22. Center for Disease Control and Prevention. Growth Chart Training. https://www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm. Accessed January 5, 2022.
23. CDISC. CDISC Study Data Tabulation Model Implementation Guide: Human Clinical Trials Version 3.2. https://www.cdisc.org/standards/foundational/sdtmig/sdtmig-v3-2. Accessed January 5, 2022.
24. CDISC. CDISC Study Data Tabulation Model Implementation Guide: Human Clinical Trials Version 3.4. https://www.cdisc.org/standards/foundational/sdtmig/sdtmig-v3-4. Accessed January 5, 2022.
25. CDIS. CDISC Analysis Data Model Implementation Guide Version 1.3. https://www.cdisc.org/standards/foundational/adam/adamig-v1-3-release-package.
26. PharmaSUG. ADaM Structures for Integration: A Preview. https://www.pharmasug.org/proceedings/2018/DS/PharmaSUG-2018-DS03.pdf. Accessed January 5, 2022.
27. U.S. Food and Drug Administration. Collection of Race and Ethnicity Data in Clinical Trials. https://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126396.pdf. Accessed January 5, 2022.
28. U.S. Food and Drug Administration. Study Data Technical Conformance Guide – Technical Specifications Documents. https://www.fda.gov/media/153632/download. Accessed January 5, 2022.