

Introduction
Historically, clinical trial sponsors monitored, including performing source data verification (SDV), all patients from whom informed consent was obtained. However, in response to revised guidance from regulatory authorities,1,2 ICH-GCP E6 (R2) Guidelines,3 and Japan-GCP,4 monitoring practice has shifted from the conventional 100% sponsor-implemented monitoring to a risk-based approach.5 Though already increasing prior, the pandemic hastened implementation of central and remote monitoring approaches. With less on-site checking by trial sponsors, activities to protect subjects and to ensure the reliability of data have increased at clinical sites and sponsor organizations, increasing the importance of process and quality management.6,7
Current industry thinking is that with RBM, data quality and patient safety can be maintained despite monitoring fewer study subjects and data elements as long as possible risks are predicted in advance and preventive measures are established preemptively. Thus, appropriate RBM practices can decrease on-site monitoring.8 In addition, the increased use of Electronic Data Capture (EDC) and electronic Patient-Reported Outcome (e-PRO) systems to review data centrally — also called central monitoring8 — reduces the frequency at which Clinical Research Associates (CRAs) must visit participating site institutions and the length of site visits. In this context, RBM practices have demonstrated improvements or non-inferiority in various aspects of quality in clinical trials as well as decreased cost.8,9,10,11 The latter is due to decreased on-site monitoring in favor of off-site (central and remote) monitoring approaches. However, the impact of RBM approaches on clinical sites has garnered less attention. To better characterize the impact of RBM practices on clinical sites, we conducted a survey of clinical research coordinators (CRC) and data managers (DM) at Japanese medical institutions.
Methods
A survey of CRCs and DMs working in university hospitals (n = 80) and cancer centers (n = 18) in Japan was conducted. In Japanese medical institutions, CRCs mainly assist in the overall operation of clinical trials and coordinate the clinical site team conducting the study. Some medical institutions also have site DMs who are mainly responsible for entering study data into the Sponsor’s EDC system. In this survey, on-site monitoring refers to the conventional approach of monitoring by visiting the institutions; whereas, off-site monitoring refers to an approach of remote monitoring via telephone and e-mail without visiting the site institutions. Remote source data verification (SDV) refers to the review and verification of data collected for the study against the source data, such as medical records, without visiting the medical institutions.
Between October 25, 2019 and November 25, 2019, CRCs and DMs working in the National Cancer Center Hospital East (NCCHE) and university hospitals and prefectural cancer centers were surveyed. The study team distributed the survey at NCCHE and asked each prefectural cancer center to distribute the survey to CRCs and DMs working at their facility. Survey response was voluntary. The survey was built and administered through web-based Microsoft Forms. In NCCHE, individuals were identifiable through email addresses used to distribute the survey, whereas individuals were not identifiable at other surveyed locations. Survey participation was voluntary.
Results
Responses were obtained from a total of 391 respondents, of whom CRCs accounted for 93% and DMs 7%. Respondents with work experience of 10 years or more accounted for the largest proportion (31%); followed by 5 to less than 10 years (29%); less than 3 years (25%); and 3 to less than 5 years (15%). Three hundred and seventy-five (96%) survey participants participated in monitoring as part of their job responsibilities. For example, participatory activities may include preparation for monitoring visits, arranging access to records for monitors, hosting monitoring visit, and responding to monitoring queries. The remaining 16 (4%) of the respondents did not perform monitoring as part of their duties.
When asked about their knowledge and experience with RBM, 40% of the 375 respondents answered, “I know RBM, but have never participated”; 37% answered, “I have been trained by the sponsor in advance, and am participating in RBM”; 11% answered, “I have not been trained by the sponsor, but am probably participating in RBM”; and 12% answered, “I don’t know about RBM.” When asked whether, if given a choice, they would choose to perform RBM, 29% answered, “Yes, I will”; 20% answered, “No, I don’t want to”; and 51% answered, “I don’t know.” Table 1 provides example text responses to the question.
Opinions from CRCs and DMs for RBM.
| Opinions from CRCs and DMs who answered that they would conduct RBM |
| Adoption of RBM will reduce the time taken to conduct SDV by CRAs and CRCs, improving efficiency. |
| Utilization of process management for preventing errors from occurring will help both medical institutions and sponsor to improve business efficiency. |
| I realize the necessity of implementing a process to reduce errors or problems during daily work. |
| Cost reduction by RBM will be reflected in drug price, eventually bringing benefits to patients |
| Opinions from CRCs and DMs who answered that they would not conduct RBM |
| A considerable change in process may be necessary because of prior risk prediction and occurrence of risks after the start of a study. |
| A difference in ability between CRAs and CRCs may cause poorly-identified risks during RBM discussion and only time and manpower may be wasted. |
| The information checked by CRAs on site so far will be reviewed by CRCs and other tasks, including creation of forms specified by the sponsor and telephone communication, will increase the burden of CRCs. |
| Delayed detection of deviation. |
| Opinions from CRCs and DMs who answered that they didn’t know that |
| It is necessary to evaluate RBM adoption by checking the target disease, study design, and other information. |
| The sponsor can reduce the visit time and costs, including transportation expenses, although the institution needs to spend more time on RBM. |
| When patient registration takes place frequently or an SAE occurs, CRAs cannot fully understand what is going on by RBM and may not be able to consult. |
| Depending on the policy chosen by the sponsor, adoption of RBM will increase the number of studies managed by each CRA, and I think the role of CRAs is underestimated. |
Current status of RBM
The survey contained more detailed questions about RBM practices and their effects at clinical sites. Of the 375 respondents indicating they participated in monitoring 182 (49%) answered either “I have been trained by the sponsor in advance, and am participating in RBM” or “I have not been trained by the sponsor, but am probably participating in RBM.” Details on the detailed survey are included in the text and figures that follow.
In comparison to traditional 100% monitoring, respondents were asked to indicate their perception regarding the relative frequency of on-site monitoring visits. Of the 182 respondents performing RBM, 51% answered, “The frequency of on-site monitoring decreased”; 30% answered, “Remains unchanged, or I don’t sense any difference”; 16% answered, “The frequency of off-site monitoring increased”; and 3% answered “other.”
These respondents were also asked, in comparison to traditional 100% monitoring, to indicate the frequency of on-site monitoring in studies using RBM. Seventy-three (40%) answered, “once in 2 months”; 44 (24%) answered, “once or twice a month”; 25 (14%) answered, “at least 3 times a month”; and 40 (22%) answered, “other”. The respondents who answered “other” were further broken down to 17 cases who answered once in 3–4 months and 5 cases who answered once in six months.
These respondents were also asked how much time they spent on each on-site monitoring visit in studies using RBM. One hundred and forty-nine (82%) indicated, “4–7 hours” per on-site monitoring visit, 18 indicated, “8 hours or more” per on-site monitoring visit, and 15 respondents indicated, “2–3 hours” per on-site monitoring visit.
Respondents were asked which RBM methods were used on their studies (studies on which the CRC or DM worked to conduct off-site monitoring in studies using RBM. The most frequently used means was e-mail — indicated by 103 respondents, followed by telephone — indicated by 93 respondents, and remote SDV — indicated by 61 respondents. Forty respondents answered, “I’ve never used any of these methods.”
Respondents were asked to freely describe the frequency of and time spent on off-site monitoring. The largest number of respondents indicated that they correspond to monitoring by telephone once every one to two months, spending 30 minutes on average per call. The range of responses was ten minutes to one hour. Many respondents stated that monitoring by e-mail is conducted after each visit where necessary. For respondents indicating the content of the monitoring, the most common responses included “concomitant medications”, “adverse events,” and “progress of EDC entry.”
Respondents were asked to state, multiple-choice and in free text, topics discussed with the sponsor prior to the start of RBM on a study. The largest number of respondents (77) indicated some aspect of or directly stated process management (Figure 1). Seventy respondents indicated that no prior discussions occurred. Fifty-two respondents indicated some aspect of or directly stated the risks relevant to each study.
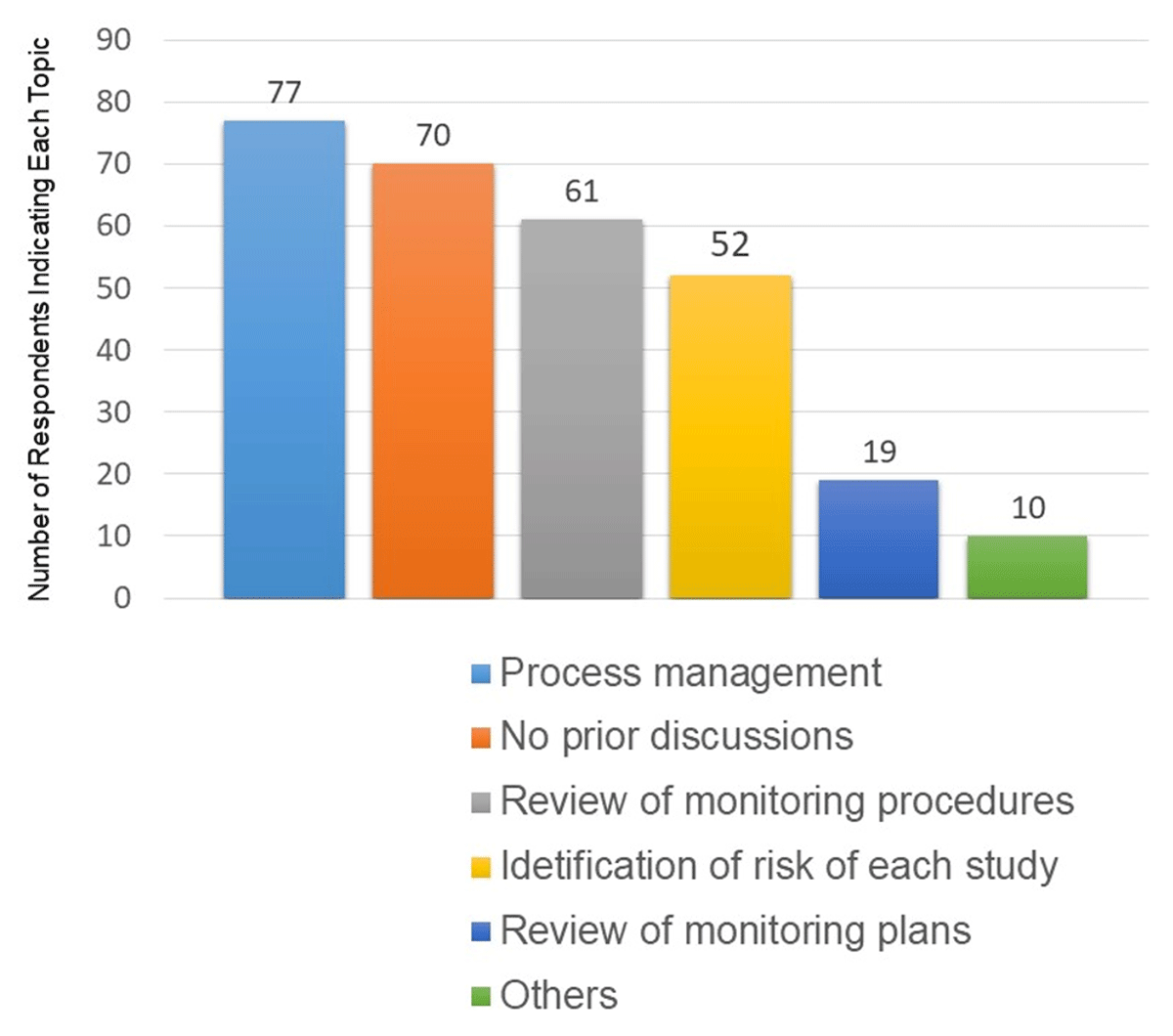
Topics discussed prior to the start of RBM.
Multiple responses were allowed in this survey question.
Respondents were asked to state, multiple-choice and in free text, matters to be discussed with the sponsor before Introduction of RBM. The question was answered by a total of 331 respondents, consisting of 182 who have performed RBM and 149 who responded, “Although I have never performed RBM, I understand it.” Free text responses to this question were allowed. The largest number of the respondents (250) answered “discussions on identification of risks for each study,” followed by two hundred of the respondents who answered, “review of monitoring procedure” (Figure 2).
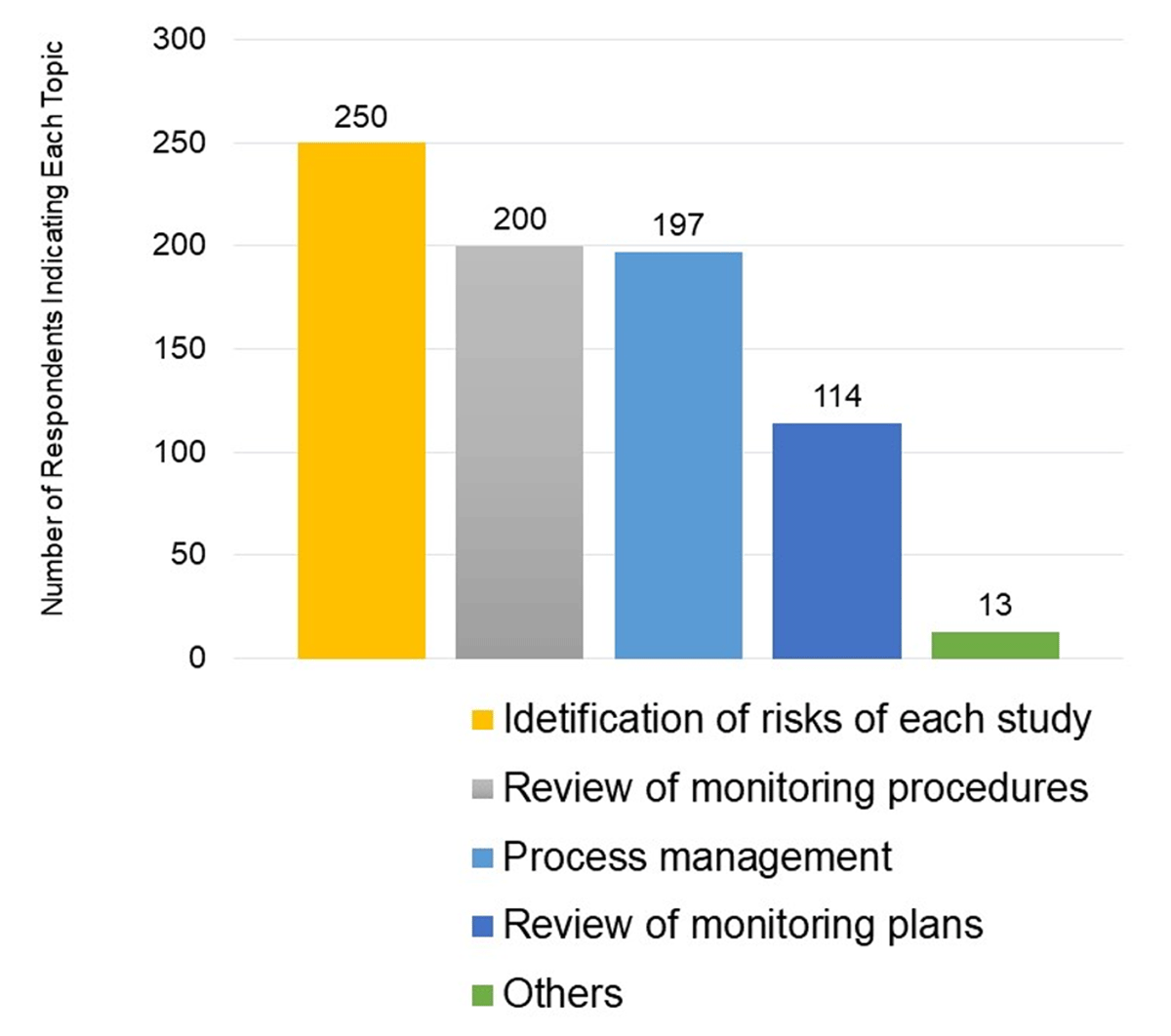
Matters to Be Discussed with the Sponsor Before Introduction of RBM.
Multiple responses were allowed in this survey question.
Items where a high risk of error is expected
The 375 respondents indicating that they participated in monitoring as part of their job responsibilities, i.e., they respond to monitoring by CRAs, were asked to state areas with a high risk of errors and that should be included in monitoring. The most frequently listed area, “eligibility/enrollment”, was stated by 311 (83%) of the respondents. Two hundred and ninety (77%) of the respondents indicated some aspect of or directly stated, “treatment suspension/dose reduction/discontinuation”, Two hundred and fifty-six (68%) of the respondents indicated some aspect of or directly stated, “initial treatment, PK and related tests, etc.” (Figure 3).
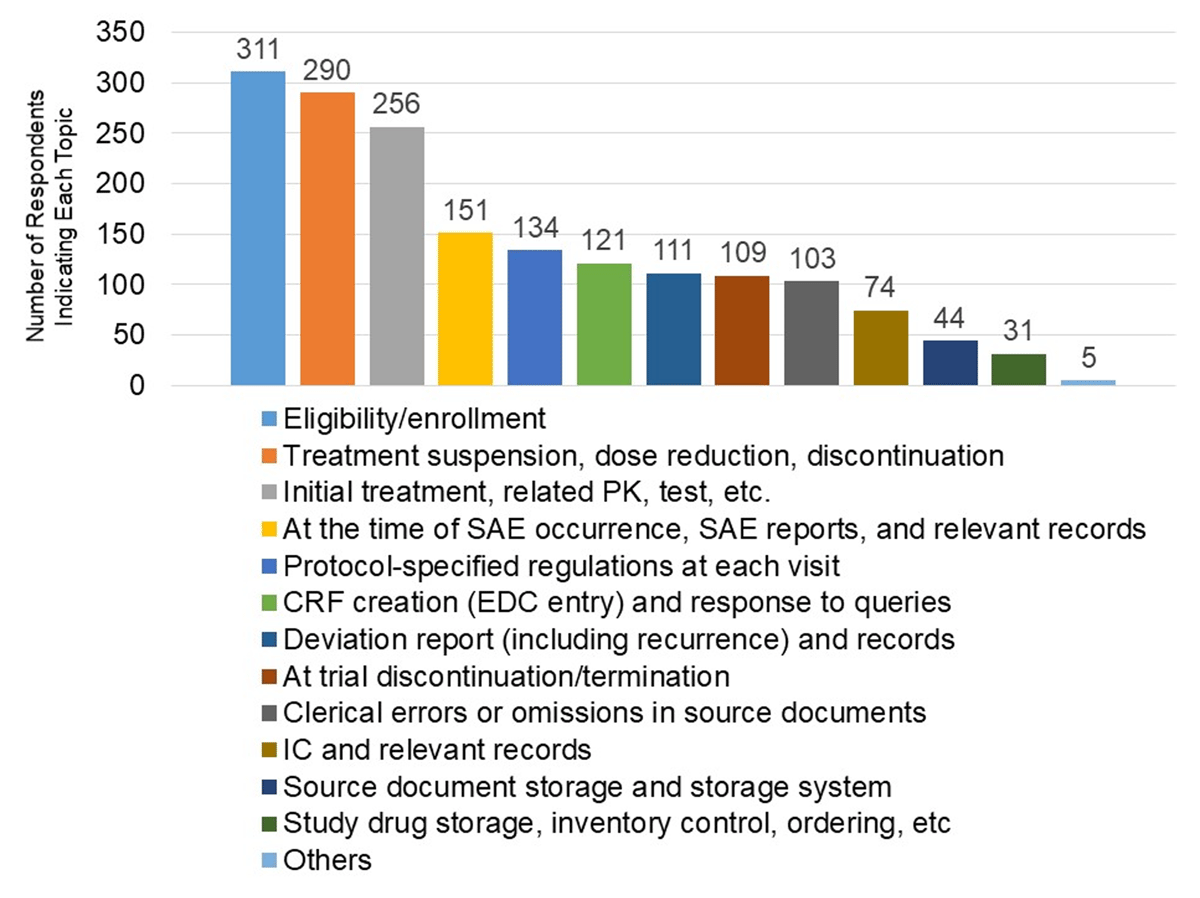
Areas Where Respondents Indicated a High Risk of Error.
Multiple responses were allowed in this survey question.
Discussion
CRCs comprised 93% of the total responses to this survey, whereas DMs were 7% (29 of 391) of the responses. This finding potentially indicates that CRCs, rather than DMs, are responsible for supporting monitoring by study Sponsors, answering questions about the data, entering data into EDC systems, and other data management duties in most Japanese medical institutions.
Introduction of RBM
For a majority of survey respondents, RBM has reduced the frequency of on-site and off-site monitoring visits by CRAs. However, there was also an opinion that increased off-site monitoring such as by telephone and e-mail resulted in an increased workload for CRCs (Table 1). The increase in workload at clinical sites is probably due to time and effort spent on the entry of the prior confirmation form (a form used for off-site monitoring is distributed by the CRA in advance), and monitors frequently requesting status reports of data entry progress from sites that could otherwise be obtained by the monitor through the EDC system. Depending on the time and length of the phone call, off-site monitoring via phone could take time from other site duties such as interacting with patients.
The method and time of off-site monitoring may vary according to the specifics of the study protocol, the stage of completion of the protocol at the site, as well as processes specific to each institution. However, we considered it important to establish process management that reflects prior risk identification and establishes a relationship based on trust between CRAs and CRCs to support effective monitoring, which is a combination of on-site and off-site monitoring.
In addition, the responses to our survey confirmed that, in some cases, CRCs and DMs took part in RBM without prior training by the sponsors. We consider prior discussions between the sponsor and the medical institution to be essential in order to identify local risks as well as risks that apply to multiple sites for individual protocols and to take preventive actions. It is important to establish a process that reflects preventive actions for risks specific to the study in the regular process of the clinical trial at each medical institution.
Traditionally, the process for quality control (ensuring subject protection and the reliability of trial results) has been established in Japan by integrating critical processes into the study data collection forms to remind or even enforce compliance with the study protocol and to enable process control. An example of a work sheet used to manage investigational product (IP) administration is provided in Table 2. Thus, sophisticated and effective mechanisms through which identified risks can be prevented or mitigated and through which unanticipated problems can be detected and resolved exist in Japan and stand ready to support RBM.
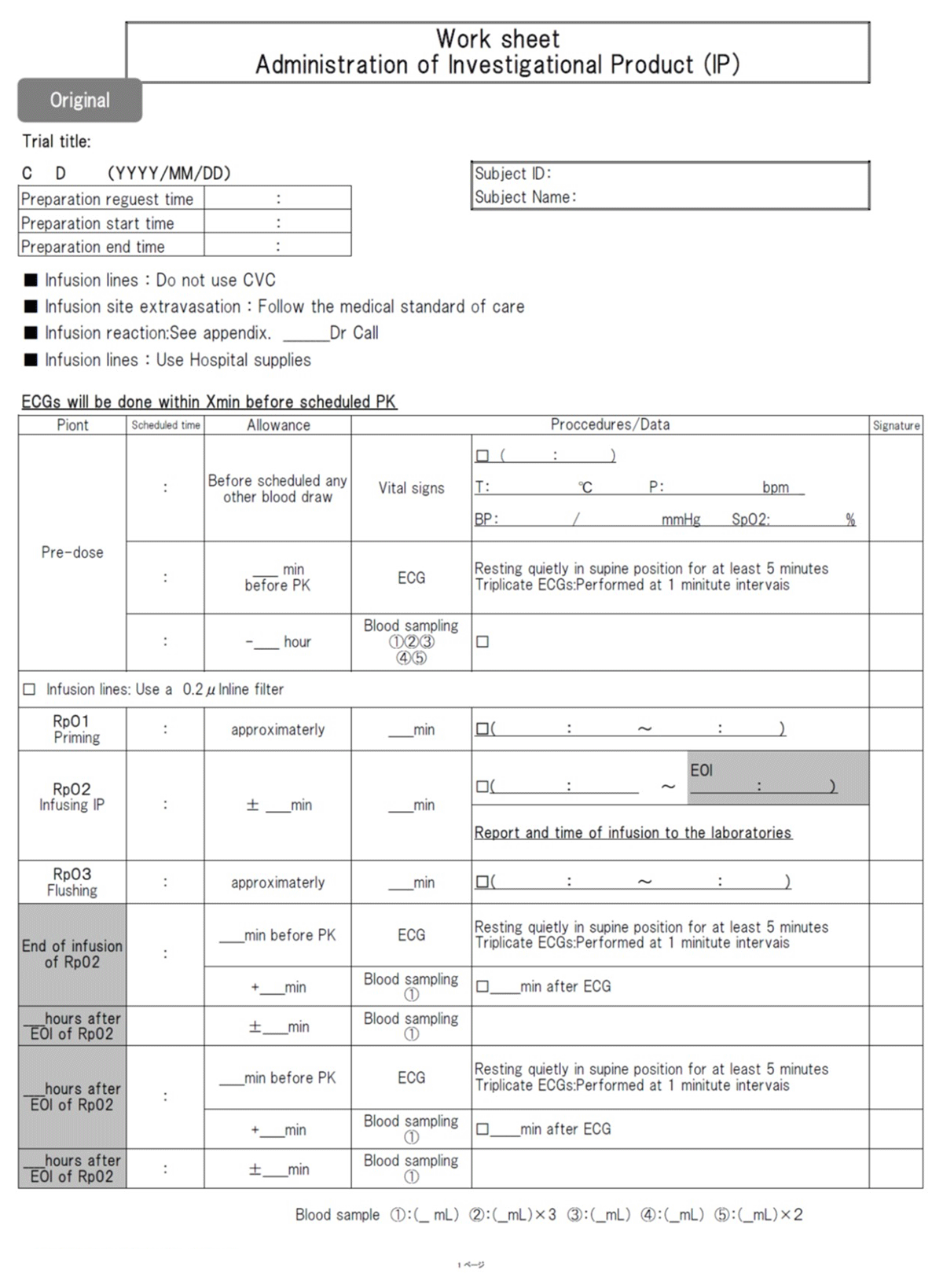
Work sheet.
Risk identification
The results of this survey indicate that the risk of error was higher in “eligibility/enrollment,” “treatment suspension/dose reduction/discontinuation,” and “initial treatment, PK and related tests, etc.” Many respondents indicated that the sponsor should include or focus on these areas in monitoring plans.
Many factors could contribute to error in these areas, including the increasing complexity of protocols in recent years leading to difficulty in sites understanding the protocol; variability in the protocol format, content, and detail among sponsors decreasing site’s ability to find information quickly and to use the protocol as a job aid; and entry of difficult or complex data into the EDC system. In addition, an error in study procedures could adversely affect a patient or jeopardize their continuation in the clinical trial. To overcome these problems, it is necessary to identify the risks specific to the protocol and to take preventive measures at the preparation stage, before the clinical trial is initiated. Further, the impact caused by the difference in experience and skills among individuals should also be taken into consideration in risk-based decisions such as the frequency, extent, and content of monitoring. In Japan, a large number of medical institutions — several dozen — usually participate in a single clinical trial. Establishing a system for sharing information, such as questions about the protocol and information on deviations, among sites not just between the sponsor and one institution, will improve work efficiency and increase data quality.
To control the quality of data in RBM, it is important for medical institutions to enter data and resolve queries more quickly than before. In central monitoring by the sponsors, our results indicate that “eligibility/enrollment,” “treatment suspension/dose reduction/discontinuation,” and “initial treatment, PK and related tests, etc.” should be intensively checked for errors and deviations and that the error rate of the data in the EDC system should be measured across all related sections of the data and in a representative sample.
Where RBM results in overall increased efficiency and decreased cost, an associated increase in off-site and remote monitoring related work at sites may be appropriate. However, such a shift in workload competes with other priorities for limited site resources and dilutes the efforts of the CRC from other trial work. Without a commensurate adjustment in resources, the work reallocation associated with RBM could itself become a risk to the trial.
Conclusion
To maximize the efficiency of RBM, the sponsor needs to optimize the use of on-site, off-site, and central monitoring practices. In particular, in terms of central monitoring, information regarding the items of “eligibility/enrollment,” “treatment suspension/dose reduction/discontinuation,” and “initial treatment, PK and related tests, etc.” should be checked intensively for errors with the information entered in the EDC system confirmed across all related sections, not just in one part of the EDC system. For example an adverse event page should be checked with related information such as dose reduction, discontinuation, or concomitant medications. In addition, to achieve the most benefit from central monitoring, medical institutions must enter data and respond to queries more quickly than previously.
Lastly, our survey indicates that an unintended consequence of RBM, especially off-site remote monitoring, is an increased workload at the clinical site. For example, faithful application of RBM requires the medical institution to participate with the Sponsor and identify local or site-specific risk for each study. Significant increases in site workload included various aspects of supporting remote monitoring. Collaboration between medical institution and Sponsors is necessity to improve efficiency through RBM.
Acknowledgements
We would like to thank both the medical institutions, CRCs, and DMs in SMO, and those involved who cooperated for this survey.
Competing Interests
The authors have no competing interests to declare.
References
1. European Medicines Agency. Reflection paper on risk based quality management in clinical trials. https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-risk-based-quality-management-clinical-trials_en.pdf. Published November 18, 2013. Accessed June 26, 2020.
2. U.S. Department of Health and Human Services Food and Drug Administration. Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring. https://www.fda.gov/media/116754/download. Published August 2013. Accessed June 26, 2020.
3. Ministry of Health, Labour and Welfare. ICH Harmonised Guideline Integrated Addendum to ICH E6 (R1): Guideline for Good Clinical Practice E6 (R2). https://www.pmda.go.jp/files/000231047.pdf. Published November 9, 2016. Accessed October 18, 2019.
4. Ministry of Health, Labour and Welfare. Guidance for the Ministerial Ordinance on Good Clinical Practice for Drugs. https://www.pmda.go.jp/files/000230974.pdf. Published July 5, 2019. Accessed October 18, 2019.
5. Ministry of Health, Labour and Welfare. Basic Concept of Monitoring System Based on Risks Associated with Clinical Trials. https://www.pmda.go.jp/files/000230972.pdf. Published July 5, 2019. Accessed October 18, 2019.
6. Ministry of Health, Labour and Welfare. Basic Concept of Quality Management in Clinical Trials. https://www.pmda.go.jp/files/000230971.pdf. Published July 5, 2019. Accessed October 18, 2019.
7. Ministry of Health, Labour and Welfare. Documents or Record of Clinical Trials. https://www.pmda.go.jp/files/000230975.pdf. Published July 5, 2019. Accessed October 18, 2019.
8. Working Group on Guidelines for Monitoring and Auditing Clinical Trials. Guidelines Concerning Monitoring and Audit of Clinical Trials. Jpn J Clin Pharmacol Ther. 2015; 46(3):133–178. https://www.jscpt.jp/press/2015/pdf/150601_all.pdf. Accessed October 18, 2019. DOI: http://doi.org/10.3999/jscpt.46.133
9. Agrafiotis DK, Lobanov VS, Farnum MA, Yang E, Ciervo J, Walega M, Baumgart A, Mackey AJ. Risk-based monitoring of clinical trials: an integrative approach. Clinical Therapeutics. 2018; 40(7): 1204–1212, DOI: http://doi.org/10.1016/j.clinthera.2018.04.020
10. Brosteanu O, Schwarz G, Houben P, Paulus U, Strenge-Hesse A, Zettelmeyer U, Schneider A, Hasenclever D. Risk-adapted monitoring is not inferior to extensive on-site monitoring: results of the ADAMON cluster-randomised study. Cinical Trials. 2017; 14(6): 584–596. DOI: http://doi.org/10.1177/1740774517724165
11. Barnes B, Stansbury N, Brown D, Garson L, Gerard G, Piccoli N, Jendrasek D, May N, Castillo V, Adelfio A, Ramirez N, McSweeney A, Berlien R, Butler PJ. Risk-based monitoring in clinical trials: past, present, and future. Ther Innov Regul Sci. 2021; 55: 899–906. DOI: http://doi.org/10.1007/s43441-021-00295-8