

Introduction
Traditional industry-sponsored clinical trial manual data entry into Electronic Data Capture (EDC) systems from the site’s Electronic Health Record (EHR) is inefficient. This process consumes valuable site and sponsor time and effort (T/E) and can introduce errors into the dataset from manual transcription processes1,2. To address these shortcomings, sites have historically worked with sponsors on point-to-point digital solutions that hasten dataset transfers3. For example, Memorial Sloan Kettering Cancer Center (MSK) launched its eSource Program in 2014 in coordination with major biopharmaceutical firms; the primary goals of the program are to enhance efficiencies, avoid redundancies, reduce errors, and decrease T/E for all parties involved. MSK learned that rapid scalability beyond a small group of biopharmaceutical firms without readily available financial and technology resources became an impediment. To address this, sites, sponsors, and technology vendors joined forces in 2017 under the Society for Clinical Data Management eSource Implementation Consortium (SCDMeSIC) to freely share best practices and move the needle forward for the sharing of available research source data through direct data exchange (DDE). The SCDMeSIC’s first area of focus was structured local laboratory data due to its volume and data maturity level4.
Background
Sponsor Case Report Form (CRF) design has been largely unchanged for the past 30 years. EDC technology advancements have transformed paper-based CRFs into online and/or cloud-based electronic CRFs (eCRFs). However, the overall data entry flow for abstraction to those systems has not changed by virtue of their transformation into an electronic format5.
The adoption of EHR systems in the U.S. has grown from <10% in 2000 to approximately 86% in 2017, particularly amongst large healthcare organizations6. With the change from paper to electronic data format in clinical research, it is desirable to explore how the EHR may be a source for improving structured clinical trial data collection and transfer. For example, Eli Lilly (Lilly) and MSK conducted a data mapping project in 2016. Using a production protocol, they identified key data elements in each eCRF page and determined what was structured and available in MSK’s source systems. Figure 1 presents the data mapping results where the vertical bars represent the eCRF data volumes by domain and availabilities, and the pie chart shows the overall eCRF data elements across the entire study protocol. Overall, 55% of data elements from all eCRF pages were available in electronic format in MSK’s EHR, including lab results (22%), adverse events (11%), and vitals (8%). A similar project was conducted by Novartis (NVS) and MSK in 2014, and they found that approximately 20% of all T/E for CRF page data entry at the site was for local lab data.
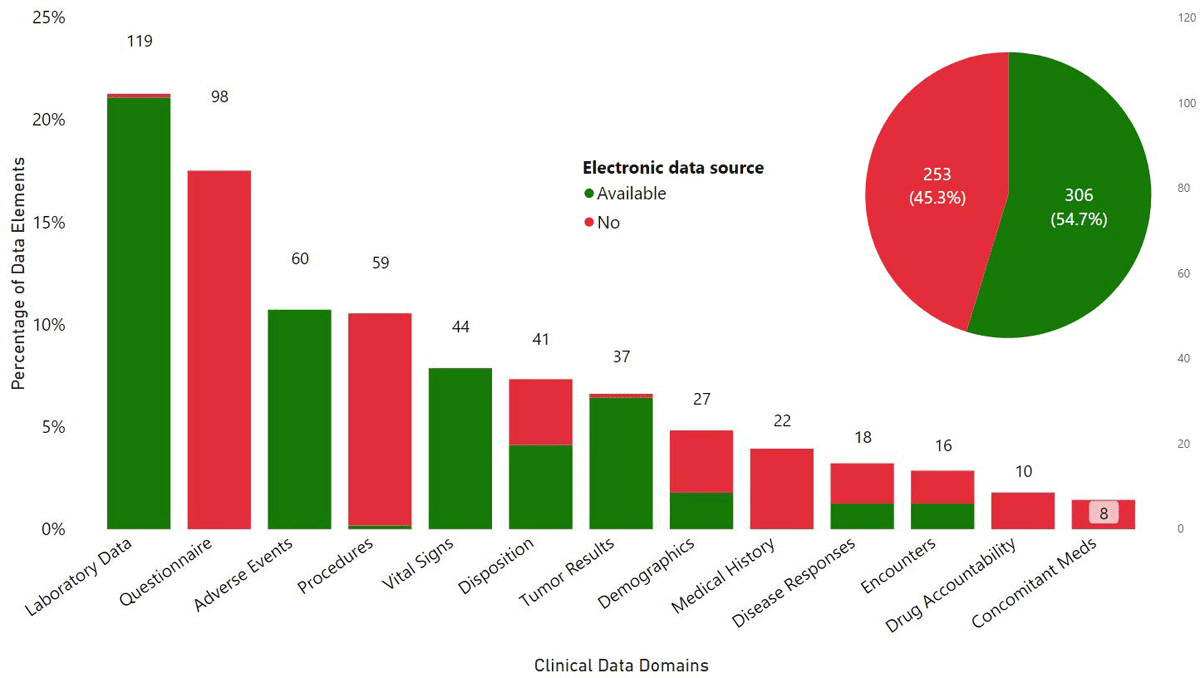
2016 eCRF structured data domain availabilities from MSK’s EHR as they relate to Lilly’s protocols.
After similar examinations regarding the structure and availability of these data domains with other SCDMeSIC member sites, local lab results stood out for both their long-standing structured format (high maturity level) and machine-generated nature (without transcription) as the ideal candidate for the initial pilot. Additionally, the patient safety nature of lab values as they relate to oncology studies was an additional driver for choosing this domain. The high volume of manual transcriptions and the need for patient safeguarding made local lab data the ideal candidate to start this journey.
Methods
Data transfer agreements form the foundation of the DDE process
Direct transfer of any data domain is codified and facilitated by the site and sponsor’s standard operating procedures (SOPs). Each clinical trial utilizing DDE has an associated data transfer specification (DTS). The DTS specifies requirements such as study-specific data elements, file format, delivery method, frequency of submission, and communication/escalation regarding any transmission/data issues.
Operationalization of DDE is enabled by a robust set of infrastructure SOPs, validation documents, and security specifications
To ensure that the DDE transfer process was codified and operationalized according to regulatory best practices for electronic data transfers, MSK created a variety of new process documentation: 1) SOPs for the use of the DDE and automation, and 2) infrastructure SOPs for the use of the DDE processes for key areas of information security, software development lifecycle, software change control, training, and software validation.
Proof of Concept (PoC) with Lilly, MSK, and Yale Medical Center (Yale)
To establish a true baseline between existing manual EDC transcription practices and the proposed DDE, a PoC comparison was conducted with Lilly, MSK, and Yale for two and a half months in 2017. Using seven Lilly oncology protocols that were in production, the MSK and Yale sites used DDE to transmit pilot data via secure file transfer protocol (sFTP) to Lilly for evaluation (see Results section). Site data management continued to manually transcribe data into Lilly’s InForm EDC for FDA submissions; however, both sites used DDE to transfer local lab data in parallel. We recorded original entry timestamps and values in the EDC, and these two site datasets were analyzed for 1) data latency, 2) transcription errors, 3) query rate, and 4) T/E savings. The processes of direct local lab data transfers from MSK and Yale (AllScripts and Epic, respectively) to Lilly’s data warehouse were developed to comply with the electronic source data regulatory framework of the U.S. Food and Drug Administration (FDA)7, the Medicines and Healthcare Products Regulatory Agency (MHRA) GxP data integrity guide8, the Health Insurance Portability and Accountability Act (HIPAA)9, and other applicable local and state laws.
Production pilot with Lilly and MSK
Backed by the PoC’s positive outcomes, a production study was selected in 2018 to further pilot the entire system from end to end, including process and change controls, source data verification (SDV) monitoring practices, scalability evaluation, and regulatory framework assessment. The same four efficacy assessment criteria noted above were also used to evaluate the pros and cons of the DDE program versus traditional manual data entry into Lilly’s EDC.
NVS’s DDE Journey with one U.S. site – MSK, and three Japanese sites – National Cancer Center East Hospital (NCCE), Shizuaka Cancer Center, and Sumida Hospital
NVS implemented DDE with MSK in 2012, NCCE in 2014, Shizuaka Cancer Center in 2017, and Sumida Hospital in 201810. DDE implementation obviated the need for these sites to enter data manually into the NVS EDC for the DDE data elements. These collaborating sites used different EHR/source systems (All Scripts, Fujitsu, and IBM) which resulted in customized site specific DDE processes.
Results
Lilly-MSK-Yale DDE PoC showed a significant productivity and Return on Investment (ROI) benefit
When compared with traditional manual transcription into the sponsor EDC (2,546 lab results), the Lilly-MSK-Yale PoC decreased data latency from 20.4 days to 3.5 days, and decreased transcription errors from 6.7% to 0% (Figure 2). DDE allowed for more efficient use of both the sponsor/Contract Research Organization (CRO) and site staff T/E. Sites reduced T/E by 8 hours per patient per study, reduced queries by 2.5 queries per patient per protocol study visit, and reduced sponsor’s monitoring activity by 3 hours per patient per study.
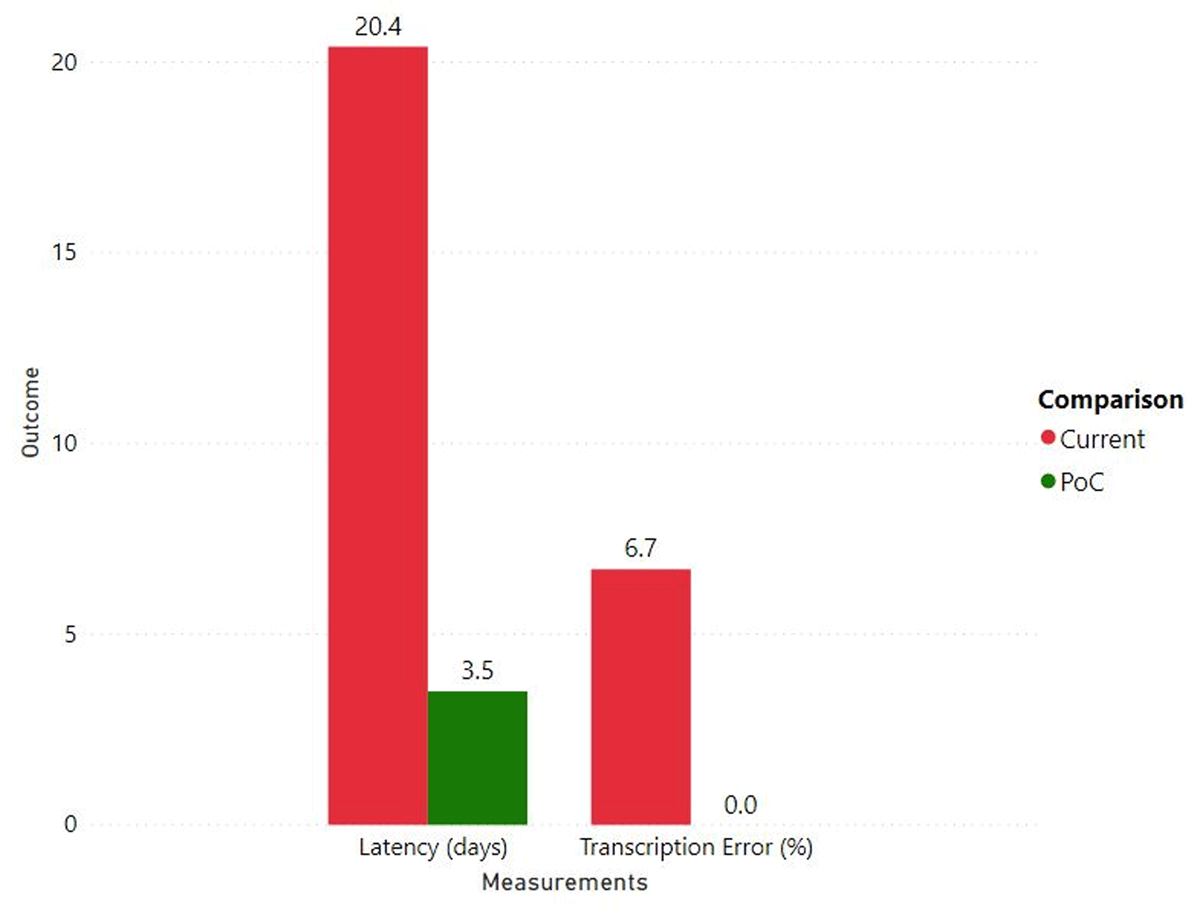
Data latency and transcription error* comparisons between current manual EDC transcription and DDE used in the Lilly-MSK-Yale PoC.
* Data latency was defined as the time from visit date to the date when all local lab data are transcribed into EDC. Transcription error was measured using the number of local lab data entry modifications in the EDC post original data entry.
NVS DDE productivity analysis with MSK and NCCE: Japan showed similar efficiency gains
A 2012 productivity analysis conducted by NVS comparing MSK’s local lab DDE with a similar site’s manual process found that approximately 20% of the effort associated with manually entered data was removed, and the number of queries was decreased by 50%. A 2015 analysis on another study determined that approximately 24% of the effort associated with manually entered data was removed when using DDE, and observed the same about 50% query reduction. A productivity analysis performed by NVS with an NCCE, Japan Phase I clinical trial with 16 patients, 148 visits, and 6,518 data points using DDE for local labs yielded the results shown in Table 1. There were two main findings: 1) reduction in 73.2 hours (99%) for NVS’ traditional data review activities, normal range population and source data verification; and 2) reduced queries to the site by 164 queries (96%)11. The other two Japanese sites, Shizuaka Cancer Center and Sumida Hospital, showed similar experiences based on qualitative feedback. No formal metrics were collected from these two additional sites, since the model was declared as a value add and a success based on two separate and independently validated use cases.
Reduced efforts (hours) and queries from the DDE transfer process compared with manual procesess in NVS-NCCE study.
| Manual data entry process | Direct transfer process | Reduction | Reduction Rate | |
|---|---|---|---|---|
| Total efforts (hours) | 73.7 | 0.5 | 73.2 | 99% |
| Queries | 170 | 6 | 164 | 96% |
The DDE program increased productivity and cost benefits for the sites and sponsors
Since 2007, MSK has used the DDE to transfer local lab data from nearly 100 protocols with approximately nine industry sponsors/CROs including Bristol Myers Squib, Lilly, and NVS. MSK has found that staff T/E was reduced 20–30% overall across all protocols using the DDE method for local labs.
Discussion
Although EHRs have been widely adopted, the structured data availability and data maturity levels vary amongst different clinical trial data domains. For example, lab results are highly structured and consistently digitized. Conversely, medical history and progress notes contain unstructured text-based content and do not lend themselves readily to the DDE process. DDE data selection and identification of available, structured data elements is predominantly site driven, and varies from site to site. For example, at MSK structured data in the lab, vital signs, and demographics domains, among others, can be obtained through DDE. Availability of structured data elements may differ from site-to-site. Until a widely adopted data standard becomes a reality across the healthcare industry, it is difficult to apply a single model across all clinical trials.
Acceptance of new processes and innovative applications in clinical research can be slow because of the unproven track records and performance standards of these methods. Concerns about security, development costs, and the acceptance of electronic source data are some of the main barriers to adoption. Different privacy practices and regulatory requirements across various countries can also hinder large scale deployment for global clinical trials.
Other barriers to DDE implementation include: 1) resistance to changes in clinical workflow, 2) variable access to the required technologies, and 3) the need for continuing software validation efforts being conducted in-house if they are not outsourced to a third-party auditor. To address these concerns, MSK’s DDE implementation framework uses a two-pronged approach: 1) robust application documentation and software validation procedures, and 2) continuous SCDMeSIC engagements with FDA and other regulatory agencies that help guide the road map and development efforts in this evolving space. MSK overcame these initial barriers to implementation by demonstrating to external auditors that their process was validated and robust. These successful system audits drove continued use of the methodology for DDE with the auditing sponsors, and enabled MSK to leverage that track record to scale to other sponsors and CROs who were hesitant to use a new process without a previously proven audit track record. We suggest that other academic sites and sponsors/CROs work together to ensure their processes are documented, validated, and reproducable. The burden for DDE system and process documentation typically resides with the site, and this is an important consideration to be aware of when moving forward with implementation.
NVS has successfully deployed DDE at scale for local labs from four sites and vitals and demography domains from one site after completing the PoCs. DDE requires point to point solutions between the site and sponsor. While DDE increases efficiencies, it is not rapidly scalable due to the T/E required at both the site and sponsor to architect a custom solution that is typically site specific.
Key success factors for DDE operationalization for sites and sponsors included: 1) shared operational and technological goals, 2) top down management support and encouragement to take mitigated operational risks with due diligence, 3) successful PoCs carried out using test data from all clinical trial phases, 4) early involvement of stakeholders from quality, regulatory, privacy, legal, and compliance offices, and 5) site and sponsor return on investment metrics showing T/E reductions and increased quality that allowed critical resources to be freed up to perform higher value clinical trial activities.
Conclusion
The recent SCDMeSIC DDE lab transfer program has enabled a faster digital exchange of clinical research source data from sites to industry sponsors. DDE has increased the productivity of an existing clinical trial data transfer process by decreasing data latency, transcription errors, and queries. DDE allows for the more efficient use of both sponsor/CRO and site staff T/E. To help scale up this approach across multiple entities from different industries, the recently announced healthcare data interoperability rules may enable future data exchange on a wider national scale12. SCDMeSIC is currently conducting feasibility projects that leverage HL7 FHIR APIs to transfer clinical trial data between sites and sponsors/CROs. The Consortium agrees that using this newer transfer technology will further reduce site and sponsor T/E and gain further data quality efficiencies.
Acknowledgements
The authors would like to thank the following, Linda King (SCDM), Rhoda Arzoomanian (Yale School of Medicine), Miyako Tanada (NVS), Kimberly O’Day, Edward Rausch and Donald Jennings (Lilly), Kristopher Kaufman and Milena Silverman (MSK).
Competing Interests
The authors have no competing interests to declare.
References
1. Donat A, Hamilton N, Khan I, Chamberlain N. The Future of Clinical Trials Using Electronic Data Capture Systems. U.S. Food and Drug Administration, Center for Devices and Radiological Health, Office of Compliance, Division of Bioresearch Monitoring. https://www.socra.org/blog/future-of-clinical-trials-using-electronic-data-capture-systems. October 23, 2018.
2. Eisenstein EL, Collins R, Cracknell BS, et al. Sensible approaches for reducing clinical trial costs. Clin Trials. 2008; 5(1): 75–84. DOI: http://doi.org/10.1177/1740774507087551
3. El Fadly A, Rance B, Lucas N, et al. Integrating clinical research with the healthcare enterprise: from the RE-USE project to the EHR4CR platform. J Biomed Inform. 2011; 44(Suppl 1): S94–S102. DOI: http://doi.org/10.1016/j.jbi.2011.07.007
4. Ruvuna F, Flores D, Mikrut B., De La Garza K, Fong S. Generalized Lab Norms for Standardizing Data from Multiple Laboratories. Drug Inf J. 2003; 37: 61–79. DOI: http://doi.org/10.1177/009286150303700109
5. Monika M. Wahi, David V. Parks, Robert C. Skeate, Steven B. Goldin. Reducing Errors from the Electronic Transcription of Data Collected on Paper Forms: A Research Data Case Study. Journal of the American Medical Informatics Asso. 2008; 15(3): 386–389. DOI: http://doi.org/10.1197/jamia.M2381
6. Office of the National Coordinator for Health Information Technology. Office-based Physician Electronic Health Record Adoption. Health IT Quick-Stat #50. https://dashboard.healthit.gov/quickstats/pages/physician-ehr-adoption-trends.php. January 2019.
7. Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry: Electronic Source Data in Clinical Investigations. September 2013. Available from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electronic-source-data-clinical-investigations.
8. Medicines & Healthcare products Regulatory Agency (MHRA). ‘GXP’ Data Integrity Guidance and Definitions. Revision 1: March 2018. Available from https://www.gov.uk/government/publications/guidance-on-gxp-data-integrity.
9. US Department of Health and Human Services. Health Insurance Portability and Accountability Act of 1996, (HIPAA) Public Law 104-191, as amended, 42 United States Code 1320-d. 1996. Available at: https://www.govinfo.gov/content/pkg/PLAW-104publ191/html/PLAW-104publ191.htm.
10. Maniar, R. Implementation of Direct Data Capture at Industry Sponsor Sites – EHR and Data Acquisition – Our Journey. Session 332 at DIA 2018 Global Annual Meeting, Boston, MA, June 26, 2018.
11. Aoyagi, Yoshihiro, Yuki Harada, Mirai Kikawa, Miyako Tanada, Nobuyuki Funami, Eri Sekine, Kyouichi Motomura, et al. Direct data transfer from HIS (Hospital information system) to Sponsor for clinical trials. Poster PO-003 at 12th Annual meeting DIA Japan 2015, Tokyo, 15–17 November 2015.
12. U.S. Department of Health & Human Services. HHS Finalizes Historic Rules to Provide Patients More Control of Their Health Data. https://www.hhs.gov/about/news/2020/03/09/hhs-finalizes-historic-rules-to-provide-patients-more-control-of-their-health-data.html. March 9, 2020.