
1.0 Introduction
People make mistakes, and this does not change in a clinical trial setting. While it is generally understood that sponsors should not have control over source records (meaning that sponsors [or delegates] should not be able to prevent changes to data), health authority regulations and guidance may result in varied interpretation, especially for specific electronic clinical outcome assessment (eCOA) data collection scenarios.
Historically, there was a tendency amongst eCOA service providers and their sponsor clients to prevent changes to eCOA data, particularly for electronic patient-reported outcome (ePRO) measures, or to allow changes to eCOA data but only after receiving sponsor approval to do so. Further, sponsors were also approving or denying data change requests (DCRs).
Over the last 10 years we have seen a steady shift in the interpretation of the regulations from inspection findings and sharing of lessons learned across sponsors and eCOA providers. If an investigator (or delegate) requests a change to eCOA data because the originator of that data (whether patient, caregiver, observer, or clinician) has noted that the original entry is incorrect, preventing the change is inappropriate. Instead, the DCR must be evaluated and implemented according to processes established for that protocol. If the DCR is refused, the regulators are likely to issue a warning to the sponsor for failure to act in accordance with the regulations, specifically for not allowing the site to maintain accurate source records. Conversely, changes made to data without careful documentation from the site can also be the source of findings during inspections and may be seen as data manipulation.
The clinical trials industry needs a scalable process for submitting, evaluating, discussing, and implementing DCRs by clinical site staff on behalf of the data originators. This publication offers an approach that:
allows investigators to maintain data accuracy, including requesting changes to data;
ensures that data changes are supported by justification that explains the context behind the request;
allows sponsor oversight over the conduct of the trial;
prevents sponsors from exercising control over source records; and
enables eCOA service providers to leverage their technology consistently, according to trial-specific processes with agreed-upon roles and responsibilities, so that the service provider is not caught in a disagreement between the site and sponsor.
These best practice recommendations enable thoughtful data changes in accordance with the original request, while also allowing an appropriate level of review, discussion, and documentation to ensure the integrity of the data.
In order to achieve that, this manuscript supports eCOA project leadership by:
highlighting key regulatory quotations that pertain to this topic with an interpretation of them and how they apply to eCOA DCRs;
establishing a set of core principles;
outlining a risk-based approach to reviewing the protocol, establishing critical data categories, ensuring sites are effectively trained, and creating tactical strategies for DCR management;
illustrating a proposed DCR process flow that is intended to be used across sponsors and eCOA service providers, agnostic to technology, to help streamline the DCR process such that sites, sponsors, and eCOA service providers can fulfill their regulatory obligations while also working together to ensure the highest quality of trial data; and
considering the trial protocol and the recommended level of documentation needed to adhere to the regulations.
eCOA data change management is consistent with Good Clinical Practice (GCP)9: Sponsors are still expected to provide oversight, guidance, and training to sites; sites are still expected to have robust documentation supporting changes; and service providers still must establish data change processes that can be followed consistently over the life of a clinical trial.
2.0 Methodology
In 2018, member firm representatives of Critical Path (C-Path) Institute’s Patient-Reported Outcome (PRO) Consortium and Electronic Patient-Reported Outcome (ePRO) Consortium collaborated on a Job Aid to provide guidelines regarding changes to PRO data in clinical trials. A presentation of the Job Aid was given at the Tenth Annual PRO Consortium Workshop in April 2019 (available at https://c-path.org/tenth-annual-patient-reported-outcome-consortium-workshop-2/). The question that was posed was whether PRO data in clinical trials should be modified or changed after the initial collection.
This presentation coincided with a shift in the clinical trial industry in the interpretation of the many guidelines and regulations from health authorities. These regulations consistently noted that investigative sites, not sponsors, were responsible for the maintenance of accurate source records, and that although responsible for oversight of the conduct of the trial in accordance with the protocol, sponsors should not exert exclusive control over source data. In other words, the question was not whether but how PRO data should be changed.
In September 2019, a formal project including 11 representatives of 7 PRO Consortium member firms, 9 representatives of 7 ePRO Consortium member firms, and 2 C-Path staff was launched to develop best practice recommendations for changing PRO data in clinical trials. The project team included sponsors, contract research organizations (CROs), and electronic clinical outcome assessment (eCOA) service providers as well as C-Path. Their mission statement was to “bring together experts across the eCOA industry to develop a best practice for industry on handling patient-reported data change requests.” Meeting monthly, the project team worked toward the goal of publishing a manuscript that would help the industry balance the investigator’s responsibility for maintaining accuracy of source data with the sponsor’s responsibility for oversight over the conduct of the trial. The project team first developed a set of core principles to guide the generation of the recommendations over the course of several months of discussion to reach consensus. Next, an outline of the manuscript was developed, and the project team divided into sub-teams to write sections of the manuscript in parallel. The sections were combined into a single draft and reviewed section-by-section during the monthly calls. If areas of disagreement arose, they were addressed through discussion by the project team to regain consensus on the processes that would be outlined in the manuscript.
Separately to the C-Path initiative the eClinical Forum held a webinar in April 2019 to discuss ePRO data changes. As a result of the feedback from the webinar, the eClinical Forum launched a formal team to look at ePRO Data Changes in June 2019. The team initially consisted of 27 members from 10 pharmaceutical companies, 4 vendors to the clinical research industry, and 1 eClinical Forum staff member. The objective of the team was to produce a white paper on ePRO/eCOA Data Changes to be shared within the industry and to be used to solicit feedback from the regulatory authorities. The team presented the work being done by the team at three eClinical Forum workshops in the autumn of 2019 (North America, Europe, and Asia Pacific), using these workshops to collect additional information and the views of eClinical Forum members not active in the team.
In spring 2020, the leaders of these three teams met and agreed to combine forces. This resulted in a 45 member team from all three groups that met every two weeks from October 2020 through November 2021. The combined team also decided to broaden the scope of the project to more generally address changes to eCOA data. In November 2021, the team completed a manuscript entitled “Best Practice Recommendations for Electronic Clinical Outcome Assessment Data Changes” that outlines best practices aligning with current health authority guidance based on expertise from all three organizations. The manuscript was peer-reviewed by members of the PRO Consortium’s ePRO Subcommittee, the ePRO Consortium (rebranded as the eCOA Consortium in January 2022), and eClinical Forum in December 2021, with 16 individuals providing feedback. This feedback was reviewed by the writing team, and the manuscript was revised between January and May 2022. Finally, the manuscript was approved by the PRO Consortium Coordinating Committee, the eCOA Consortium Coordinating Committee, and eClinical Forum’s Steering Committee in July 2022 before being submitted for publication.
3.0 Applicable Regulatory Standards
Regulations and guidance documents create a consistent and comprehensive set of standards regarding the roles and responsibilities of various stakeholders with respect to eCOA data changes. Relevant regulations and guidelines include (but are not limited to):
FDA 21CFR312.62 (b) “Investigator recordkeeping and record retention. Case Histories”1
FDA 21CFR312.50 General responsibilities of sponsors1
European Medicines Agency. Guideline for good clinical practice E6(R2)2
FDA Final Guidance “Electronic Source Data in Clinical Investigations, 2013, Data Capture, 4 Modifications and Corrections”3
The EMA Reflection Paper on eSource4
FDA Final Guidance “Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims”5
The DRAFT EMA Guideline on Computerised Systems and Electronic Data in Clinical Trials6
CDISC Glossary, 2020-12-187
Center for Drug Evaluation NMPA Guiding Principles for the Application of Patient-Reported Outcomes in Drug Clinical Development (Trial)8
The following is a list (Table 1) of the excerpts of regulations that apply to eSource data changes and oversight that are relevant to these best practice recommendations.
Regulatory Text.
| “An investigator is required to prepare and maintain adequate and accurate case histories_that record all observations and other data pertinent to the investigation on each individual…”1 |
| “Sponsors are responsible for selecting qualified investigators, providing them with the information they need to conduct an investigation properly, ensuring proper monitoring of the investigation(s), ensuring that the investigation(s) is conducted in accordance with the general investigational plan and protocols contained in the IND…”1 |
| “Only a clinical investigator(s) or delegated clinical study staff should perform modifications or corrections to eCRF data.”3 |
| ICH E6 R2, 5.0.1: “During protocol development, the sponsor should identify those processes and data that are critical to ensure human subject protection and the reliability of trial results.”2 |
| ICH E6 R2, 6.4.9: “The identification of any data to be recorded directly on the CRFs (i.e., no prior written or electronic record of data), and to be considered to be source data.”2 |
| The EMA Reflection Paper on eSource at Topic 2: Creation, modification and transfer of data: “The location of source documents and the associated source data should be clearly identified at all points within the capture process (Requirement 11, ICH GCP 6.4.9).4 The protocol should identify any data to be recorded directly into the CRFs that is considered to be source data. A detailed diagram and description of the transmission of electronic data should be provided in the protocol. The source data and their respective capture methods should be clearly defined prior to trial recruitment (i.e., in the protocol or study specific source data agreement). The sponsor should describe which data will be transferred, the origin and destination of the data, the parties with access to the transferred data, the timing of the transfer and any actions that may be triggered by real-time review of those data.”4 |
| “A procedure should be in place to address the situation when a study subject or other operator capturing data, realizes that he/she has made a mistake and wants to correct the recorded data.”4 |
| “The investigator should maintain the original source document or a certified copy.” (Requirement 5, ICH GCP 2.11, 5.15.1)4 |
| “Source data should only be modified with the knowledge or approval of the investigator.” (Requirement 6, ICH GCP 4.9.3, 4.9.4 and chapter 8)4 |
| “The sponsor should not have exclusive control of a source document.” (Requirement 10, ICH GCP 8.3.13)4 |
| “Any change or correction to a CRF should be dated, initialed, and explained (if necessary) and should not obscure the original entry (i.e., an audit trail should be maintained); this applies to both written and electronic changes or corrections (see 5.18.4(n)).2 Sponsors should provide guidance to investigators and/or the investigators’ designated representatives on making such corrections. Sponsors should have written procedures to assure that changes or corrections in CRFs made by sponsor’s designated representatives are documented, are necessary, and are endorsed by the investigator. The investigator should retain records of the changes and corrections.” (ICH GCP 4.9.3)9 |
| “Sponsors also should avoid the following:” “Source document control by the sponsor exclusively.”5 |
| “Clinical investigator inability to maintain and confirm electronic PRO data accuracy. The data maintained by the clinical investigator should include an audit trail to capture any changes made to the electronic PRO data at any point in time after it leaves the patient’s electronic device.”5 |
| “The protocol should identify any data to be recorded directly into the eCRFs and considered to be source data (ICH-GCP 6.4.9). This is equally applicable to other specific data collection systems, such as ePRO. Data directly captured in these tools without prior identification in the protocol to be source data is considered as GCP-noncompliant.”6 |
| “… a procedure should be in place to address the situation when a data originator (e.g., investigator or trial participant) realizes that she/he has submitted incorrect data by mistake and wants to correct the recorded data.”6 |
| “Data changes for ePRO typically differ from that of other EDC tools because trial participants may not have access to correct data in the application. Hence, procedures need to be in place in order to implement changes when needed. This could be in the form of data clarification processes initiated by trial participants on their own reported data or initiated by investigators.”6 |
| “Data reported should always be reliable and it is not acceptable that data clarification procedures introduced by the sponsor or vendor whether or not described in the protocol do not allow for changes in trial participant data when justified e.g., if the trial participant realizes that data has not been entered correctly.”6 |
| “It is expected that the possibility for changes is implemented based on a justified and trial-specific risk-assessment and that any changes are initiated in a timely manner by the participant or site staff and in case of the latter is based on solid source at investigator sites, e.g., phone notes or emails from trial participants documenting the communication between sites and trial participants immediately after the error was made/discovered. Preferably such notes should be signed by the trial participant to avoid that sites are manipulating data, e.g., to make participants/patients eligible for trial participation.”6 |
| “One of the advantages of direct data entry by the trial participant is that recall bias is minimized as the data are entered contemporaneously. Consequently, corrections should not be done at a much later stage without good reason and justification. Whether collected by paper or electronic means, the regulatory requirements are that all clinical data should be accurately reported and should be verifiable in relation to clinical trials.”6 |
| “It is expected that the amount of changes to ePRO data is limited; however, this requires both designs of ePROs that are appropriate to ensure proper understanding by trial participants and appropriate training of trial participants, thereby avoiding entry errors.”6 |
Maintaining the integrity and accuracy of eCOA source records is the responsibility of the investigator and not the sponsor, and there is no distinction in regulations or guidance between patient-entered records versus other source records. Sponsors should provide comprehensive guidance to investigators on making corrections and should have written procedures to ensure changes made by the designated representative are necessary, are endorsed by the investigator, and are documented. Sponsors must not directly make changes to source records. Such changes are to be made only by a clinical investigator or authorized delegates. Sponsors are required to oversee site compliance with the protocol, and for significant non-compliance, sponsors may terminate investigator participation. Thus, to the extent that a sponsor wants to ensure procedures are in place to minimize changes to specific types of eCOA data, such procedures could be specified in the protocol or protocol-linked document. Sponsors have responsibility for the quality and reliability of the trial data they receive, which are drawn from source records, and therefore should have procedures in place to monitor when changes are made to eCOA source data to inform how these data will be analyzed and reported.
The regulations and guidance were developed by multiple regulatory bodies for application in different settings, and their application can often be challenging in situations where data modifications directly impact the outcome associated with that data point. This issue is compounded further when revisions to data impact other critical trial outcomes, which is possible in scenarios where these data are used to determine trial eligibility or to support key endpoint(s). Furthermore, these regulations do not provide comprehensive guidance regarding the appropriateness of modifications to specific types of eCOA data, especially those that are inconsistent with the development and administration guidelines associated with licensed measures.
4.0 Core Principles and Best Practice Recommendations
When designing clinical trials and associated data collection systems, it is difficult to predict all unique data change scenarios that may arise within the day-to-day execution of the trial. Since eCOA source data are collected electronically directly from the site, trial participant, or observer, often with no other source available or required, it is essential that everyone involved follows a core set of guiding principles to maintain the integrity of the clinical trial data:
Clinical data values should always reflect the respondent’s chosen response without bias or interpretation by a third party.
The clinical data value should be recorded in a manner consistent with the user guide and administration instructions of the assessment, including the recall period, if specified. If the revised value could be subject to sources of bias that call into question the validity and integrity of the data, additional actions may be required, as outlined in subsequent sections.
Data changes should not compromise compliance with Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring and Available (ALCOA+) principles.4
All data changes should be fully recorded in the system(s) audit trail to enable the site to have access to the complete eSource record and to allow the sponsor to determine and document the entries to be included in statistical analyses.
Sponsors are responsible for oversight of the service provider, including development of an oversight plan (e.g., Data Management Plan [DMP]) that will outline all data management processes to be followed for the trial. Critically, the oversight plan should address the following:
Identification of critical data points (defined in section titled: Trial Set-Up: The Oversight Plan), as well as procedural data points.
Expectations for the investigative site around the DCR process and documentation requirements. Sites must be responsible for maintaining accurate eCOA records in a compliant manner and in accordance with the protocol.
The process on when and how to notify the sponsor (and/or CRO), at the appropriate time, such as upon receipt/completion of the DCR. When appropriate, sponsors should have visibility to source DCRs in a timely manner to provide guidance, for example, at the time that the service provider receives the DCR.
Sponsors have the responsibility to train sites and to monitor, evaluate, and provide guidance on DCRs that could impact trial protocol compliance, participant safety or privacy, device functionality, or data integrity.
Sites are expected to submit DCRs related to clinical data when discrepancies are found; if sponsor (or delegate) users submit DCRs on behalf of sites, site authorization for each request is required. Changes to system data or meta-data related to performance of the eCOA system may occur and would be handled independently of this workflow but should be added to the oversight plan.
DCRs should be supported by documentation to reconstruct the eCOA data events, including the site personnel who requested and approved the change, date and time of change, and justification for change request as agreed upon by the data originator at the time the change was requested.
The core principles are the foundation that are woven into the rest of this publication to ensure sponsors, investigators, and eCOA providers can build and agree upon a process when working together on DCRs.
4.1 Trial Set-Up: Minimize Potential User Errors
eCOA systems can be comprised of different technologies for data capture. These can include devices such as a smartphone, a tablet, or a web browser/app. There may be one device or multiple devices for collection of eCOA data throughout the trial. This is dependent on the types of measures that are being completed, the disease area, the patient population, and the region(s) in which the trial will be conducted. To minimize potential DCRs, participant usability16 should be considered when selecting the mode(s) of data collection, including the use of contingency plans covering such issues as technology disruption or the ability of participants to travel to the site (i.e., pandemic).
To minimize potential errors, several considerations should be kept in mind when implementing an eCOA system. Prior to beginning the design phase, the service provider should thoroughly review the protocol with the sponsor to ensure that all requirements for the trial data to be collected in the eCOA system are documented. The critical sections should include, but are not limited to, the data collection schedule, measures and, if applicable, eligibility requirements. In addition, edit checks and other rules (such as minimizing scrolling12) can be programmed to help minimize user error. Lastly, a user acceptance testing (UAT10,15,17) process should be conducted by the sponsor or designee to simulate various test cases of eCOA data collection. Documentation of the UAT10,15,17 process and results must demonstrate accurate and satisfactory performance of the eCOA system prior to the enrollment of the first participant.
4.1.1 Trial Set-Up: The Oversight Plan
Our objective is to balance sponsor oversight of the conduct of the trial with site control over source records. To achieve this balance, a process is required that keeps sponsors informed of changes to critical data points, but also allows sites to request changes to data that were entered incorrectly, and that are required per discussion with the data originator.
A data change strategy (documented within an oversight plan, such as a DMP or similar document) should be developed and implemented for each protocol with the expected rules of engagement for each type of DCR, including requests to change critical data points. For the purposes of this paper, the term “oversight plan” will be utilized.
The oversight plan will define procedural and technical details related to each type of change. It will also define which DCR types are critical (changes that require follow-up with the site, notification/discussion with the sponsor/CRO, or both), and which types are procedural (changes that do not require follow-up).
It is recommended to start by categorizing the protocol data into critical data points and procedural data points, and then documenting these in an oversight plan. Critical data points are defined at the trial level prior to the start of the trial. All trials will differ, and the following characteristics can inform the decision to deem data as critical:
data changes that may affect participant safety;
patient- or caregiver-entered data (i.e., PRO or ObsRO data) that are deemed critical to the objectives of the trial;
primary or secondary trial objectives;
data points that affect calculations impacting specific timepoints in the trial, such as randomization, inclusion/exclusion criteria, withdrawal, entry into extension period, responder status determining ongoing treatment or dosage changes, or data points that are used to derive or calculate other such data points;
changes to date or timestamps captured by the device; and
branching functionality of an eCOA device design (e.g., changing of the data point would impact the collection of assessments A, B, C vs. only assessment A).
The oversight plan, by categorizing trial data into critical and procedural groups, would then further establish for each data type what process should be followed in the implementation of the data change. (See Appendix 2 for an example oversight plan related to eCOA DCRs.)
Control over eSource records resides with the investigator and the investigator should have access to all eSource records and potential/pending data changes.11 The sponsor is responsible for oversight over the conduct of the trial. The oversight plan should establish those data change types that may require notification to the sponsor and that may also require confirmation from the site that the rationale for the change is documented, such as changes to critical data. The purpose of any notification to the sponsor related to DCRs is informational. This notification does not constitute a request for sponsor approval but may warrant sponsor action or a pause in the processing of the DCR may be necessary to ensure the appropriate stakeholders are allowed ample time to evaluate the request. The request for confirmation from the site that the rationale for the change is documented is meant to ensure that any conversation with the data originator is adequately documented, including the date and time of the conversation and the particulars of the data entry error. The service provider and sponsor should agree in advance on the process to be followed for changes to all data types and should document these processes in the oversight plan, including any specific language to be used in the notification to the sponsor or in the query to the site.
The oversight plan may also call out other special instructions, based on data type, and specific features of the protocol that may require attention. For example, in the case of patient-reported data where the recall period of the measure is important, the site should document in the DCR the date on which the originator of the data realized the error. If the change is outside of the defined window or moves the date outside the applicable recall period, this may impact the sponsor’s analysis of the data.
Although we have focused in this publication on site requests to update clinical data, we also recognize that there are types of data changes that do not strictly fit into this process. The technology that supports eCOA data in clinical research may require updates to meta-data or system data to ensure expected device functionality or accurate data reporting. These changes may be assessed and made by the service provider without requiring sites to request the changes explicitly; however, the data changes made as part of such a process should be communicated to both the sponsor and the site and be documented in such a way as to help reconstruct the root cause and the investigation that led to the correction of the data.
The oversight plan should also outline the process for each type of data change in a way that is consistent with the protocol and maintains data integrity, and which also satisfies the regulations regarding site control over source records and sponsor oversight of the trial.
4.1.2 Trial Set-Up: Training Site Staff
The goal of training on eCOA collection best practices potentially mitigates downstream impacts on the data and ideally reduces the number of DCRs submitted. It is important to train site staff in advance on eCOA data collection best practices including DCR guidelines before DCRs occur. Considerations for training13 include the following:
Ensure that sites, participants, and, if applicable, caregivers are trained to understand the measures and their use prior to initial data collection.
Provide in-depth training15 on the DCR process separate from the investigator meeting, for example at the site initiation visit. The focus of such training should be just-in-time, not only introduced at the investigator meeting(s), but revisited once participants are enrolled. Provide manuals or other documentation for reference.
Demonstrate how the eCOA system, dataflow, and eCOA data collection devices/applications work.
If needed, describe how to troubleshoot devices, as well as how to obtain technical support to minimize DCRs.
Due to the increasing potential for complexity in the design of eCOA studies, training plays a critical role in not only preventing mistakes that would lead to DCRs, but also in ensuring that mistakes are communicated quickly to the service provider, thus mitigating downstream effects of user errors, such as missing data,14 incorrect calculations, or data captured at invalid time points. This will also help to make the user experience (whether clinician or participant) as smooth as possible.
4.2 Data Change Process Flow
The site is responsible for collecting and maintaining the clinical trial data, and the sponsor is responsible for oversight of that process. Only a very small percentage of eCOA data points ever become the target of a DCR, but this can still be a significant number of DCRs in a large trial. Figure 1 provides a generalized workflow diagram outlining the process of review and implementation of DCRs.
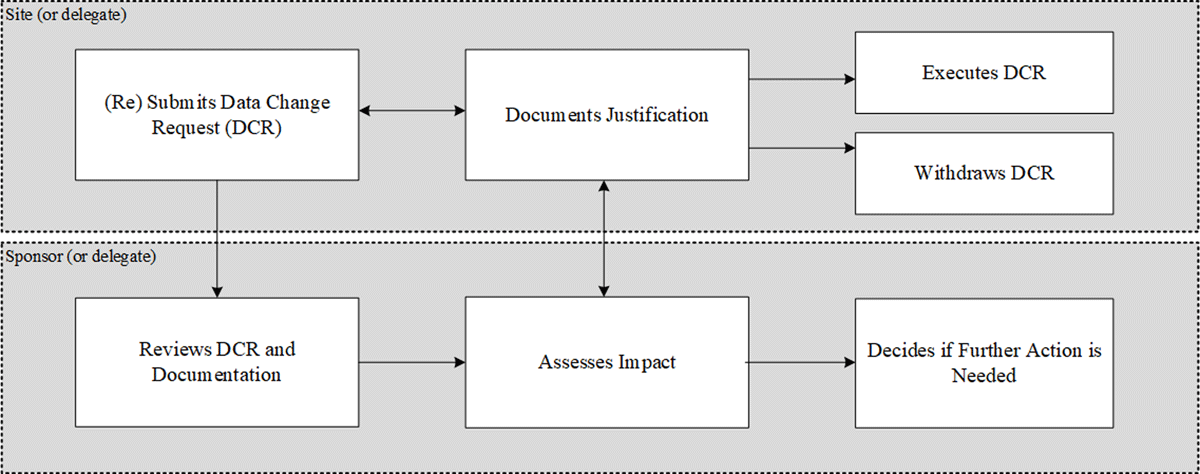
DCR General Process Flow.
As can be seen in Figure 1, the process starts when the site submits a DCR. The site then ensures that all necessary source documentation is updated with the justification for the DCR. In parallel, the sponsor or delegate will be checking the DCR based on the oversight plan. If critical data are the target of the DCR, the sponsor or delegate should review the updated source documentation. If at this point the sponsor does not think the DCR should be executed, they can discuss this with the site and if the site agrees with the sponsor, then the site can withdraw the DCR. If, on the other hand, the site insists on executing the DCR, the sponsor must then reflect on how this will affect the trial and decide whether they should take action as a result (e.g., use data analytics to determine the impact and extent of data changes; audit the site; stop recruitment within that site; report to ethics committee; exclude the data in question from the analysis dataset or other action, as applicable).
Executed DCRs controlled by this workflow are supported by:
relevant site source documentation (which is maintained at the site) and
sponsor documentation to substantiate decisions (if applicable) on inclusion of data or follow up with site.
The roles and responsibilities involved in the DCR process will differ depending on the systems used and the trial design. The execution of the change may be a manual update performed under a controlled process or it may be one that is executed automatically within a validated system. While some platforms require that data changes be made on the “back-end” by a data administrator (or specialized user type), in other platforms changes can be made directly in the front end of the system. In these “front-end” systems, specialized user types can be configured and validated to control the data change workflow and ensure alignment with the oversight plan. Each eCOA service provider leverages various technical solutions to enable data changes to be requested, tracked, implemented, checked, and reported on. Each provider, therefore, will not necessarily support the same workflow.
A common scenario among systems used in the last 20 years is the case of an eCOA provider whose own staff execute DCRs by making “back-end” changes; see the adapted workflow in Figure 2. Figure 2 is an example of a trial and system-specific workflow which should be documented in the oversight plan.
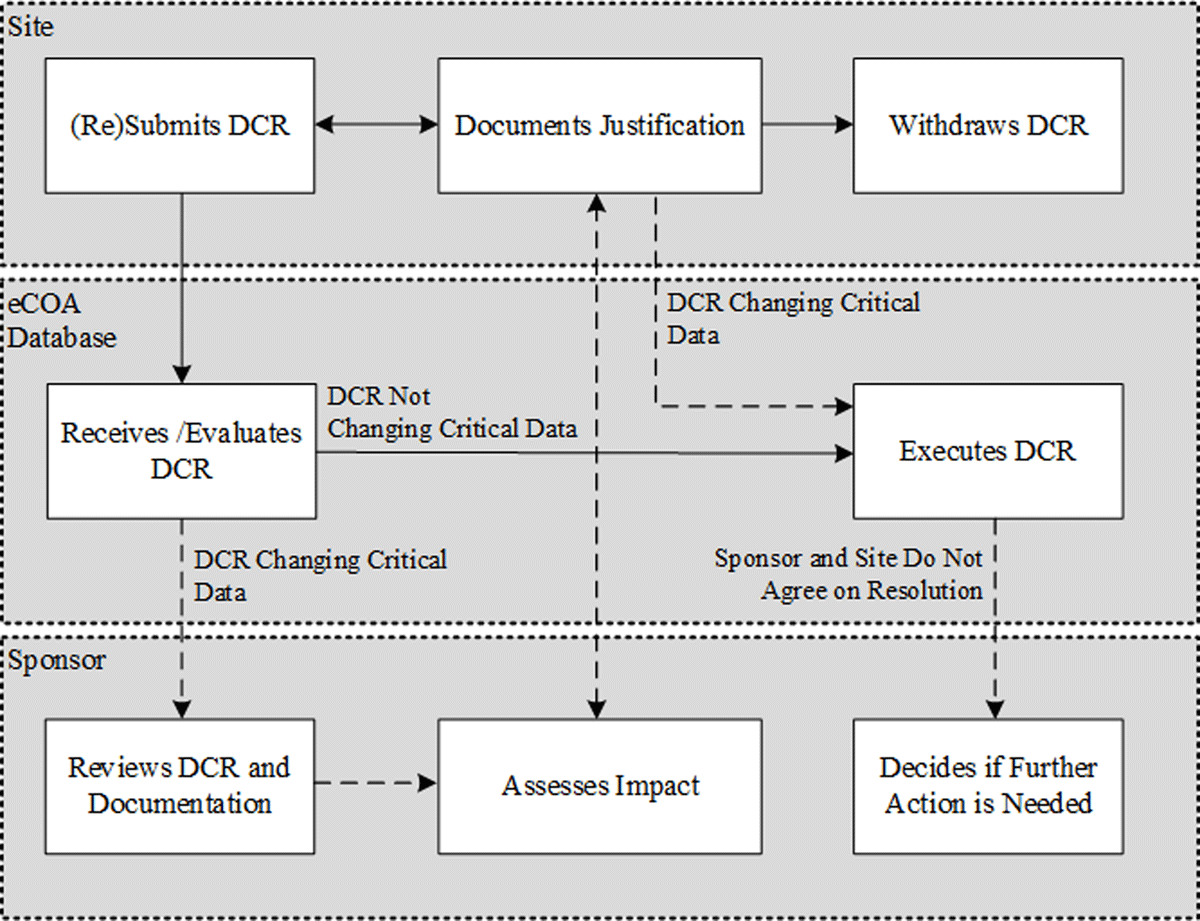
DCR Trial Specific Process Flow.
4.2.1 Standard workflow: Change type is defined in oversight plan and is procedural
DCR is submitted by site to the eCOA provider.
eCOA provider reviews DCR per guidelines established in the oversight plan.
As the DCR requests a change that is defined in the oversight plan and is a procedural data point, the change is executed.
4.2.2 Alternate workflow: Change type is not defined in oversight plan or is a critical data point
DCR is submitted by site to the eCOA provider.
As the DCR is of a critical data point, additional steps (for example, to query the site for additional information) may be required to assess the impact of the data change before execution. These steps should be pre-defined in the oversight plan, but if they are not clearly defined in advance, additional input from the sponsor may be required. After the sponsor has had the opportunity to discuss a specific DCR that affects critical data with the site, there are multiple possible outcomes depending on the protocol requirements, which may include any of the following, so as not to incorrectly influence the site:
The investigative site confirms that the relevant supportive documentation is on file and provides the context and rationale for the change, and the change is implemented; or
The site withdraws the request after recognizing that the relevant evidence is not on file, or the change is not in accordance with the protocol; or
The site and sponsor do not agree on the same resolution. The site confirms the necessity of the change, and the request is processed. The sponsor does not have the authority to deny the change but may take action as a result (audit the site; stop recruitment within that site; report to ethics committee; exclude the data in question from the analysis dataset).
Executed DCRs controlled by this adapted workflow are supported by eCOA provider system/data management processes and notifications to site and sponsor of the change by the eCOA provider.
4.2.3 Table 2 is a Summary and comparison of the 2 common DCR models
DCR process within the 2 common models of data change systems.
| Step | Back-end “Manual” Data Change System | Front-end “Automated” Data Change Portal |
|---|---|---|
| 1 | Site personnel (investigator or delegate) initiates a DCR. | |
| 2a | If the request is to change procedural data, the DCR is processed as is. | |
| 2b | If the request is to change critical data (as defined in the oversight plan), the service provider personnel may notify the sponsor, ask the site to confirm the context and rationale behind the request, or both. In a back-end system, these steps may have to be performed manually (email, query to site). | If the request is to change critical data (as defined in the oversight plan), a sub-workflow may be initiated whereby the sponsor or CRO is notified of the request and the site is prompted to confirm within the system that the change is supported by justification. |
| 3 | The sponsor may reach out to the site, directly or through the CRO, to determine the context and rationale for the request. The site may elect to withdraw or retain the request after this discussion. | |
| 4 | In the back-end system, the service provider implements the change once the site responds to the query posted from the DCR confirming that the change is required and that the rationale behind the change or corresponding source notes are in place. | In the front-end system, the DCR is processed upon receipt of confirmation from the site that the change is necessary and that any conversation with the participant is documented. |
| 5 | The DCR is marked as completed. | |
| 6 | Should the site recognize after the DCR is processed that the change is either not warranted or that the conversation with the participant has not been properly documented, the site may submit a new DCR to revert to the original values. | |
-
DCR: data change request; CRO: contract research organization.
4.3 Trial Documentation
The protocol and associated training documents relating to the protocol (e.g., investigator meeting materials) should describe appropriate management of source data indicating details to be documented within the oversight plan. Since sites do not receive the oversight plan, the protocol or other site-facing documents (e.g., those used in site training) need to clearly outline the sites’ responsibilities.
Example of generic protocol/training language:
The author of an entry in the source documents should be identifiable given that PRO responses come directly from the participant, without interpretation by a clinician or anyone else. Any changes to the original entries must be supported by a timely and adequate explanation. Timely means as soon as possible after the error is identified, and to be adequate the explanation of why the original value was incorrect should include:
the site personnel who requested the change;
the date and time the change was requested; and
the justification for the change as agreed upon by the data originator.
The change, together with evidence supporting the explanation, must be consistent with ALCOA+ principles. Specific details that help to explain or contextualize DCRs should be maintained as part of the eSource record, in accordance with ALCOA+ principles, to allow reconstruction of the events that led to the change request. The evidence recorded should be detailed given that inspectors and auditors are concerned with data integrity.4 There is a risk to the integrity of the trial data when evidence for changes to critical data points is missing or inadequate.
Any discussion between the investigative site and the data originator that results in a request to change critical data should be documented to enable the rationale and context for the change to be reconstructed. If this change is implemented, the DCR audit trail will act as the reference to the source history.
4.4 Sponsors’ Responsibilities in Reviewing DCRs
Sponsors should have the opportunity to review DCRs in a timely manner along with the site documentation as part of the monitoring strategy. It is important to note that sponsor review should not constitute an approval step within the data change process; it is the investigator who maintains control over source records. The review process implemented by sponsors should be outlined within trial documentation (e.g., monitoring plan) to ensure the sponsors are taking responsibility for oversight of the clinical trial. Sponsors or designees should verify documentation pertaining to the change and ensure adequate rationale has been provided within the DCR to reconstruct the eCOA data events, including a record of the site personnel who requested and approved the change, date and time of change, and justification for change request as communicated by the data originator at the time the change was requested.
If a sponsor disagrees with a site-initiated data change request, based on current regulations, they cannot prohibit the change, but they can discuss their concerns with the investigative site. If the site decides to reverse or withdraw the request, it is at the discretion of the site. If the change is implemented, the sponsor can decide whether the modified data should be excluded from the final analysis either by flagging the data in the data transfer, if possible, or by documenting the concern in their data analysis log, to allow possible sensitivity analysis of the results with and without the modified data.
5.0 Conclusion
Historically, service providers within the eCOA industry have approached changes to eSource data in different ways. For some providers, the agreement with the sponsor was that “Changes to patient-entered data are not allowed.” Depending on the phase, therapeutic area, and trial design, changes may have been allowed only if approved beforehand by the sponsor.
The regulations divide responsibilities between site and sponsor, where the former should have control over the source records and the maintenance of those records, and the latter should have oversight over the conduct of the trial in accordance with the protocol.
The lack of a best practice for eCOA data changes has created challenges for participants, site personnel, clinical research associates, service providers, and sponsors. Each stakeholder works to fulfill their own obligations within the responsibility of the protocol, GCP, and the regulations, but the edges of best practice have not been clear.
In this best practice recommendations article, we have described a workflow that mitigates these issues and allows for site control over source data as well as sponsor oversight over the conduct of the clinical trial. In this workflow, the site has fulfilled its regulatory obligations; the service provider has acted on behalf of the site (in making the change) and on behalf of the sponsor (in notifying them of the request). The sponsor does not have control over the source data, but instead is provided with timely information that may help explain the need for re-training of site staff or trial participants. The audit trail preserves the original and corrected values. The DCR contains the site’s explanation and/or documentation supporting the request and preserves the intention of the data originator. This article meets the need for a well-defined, consistent, and balanced process for handling eCOA DCRs in clinical trials that will result in improved data accuracy and integrity, greater transparency among the different stakeholders, increased consistency throughout the clinical trial industry, and better adherence to the trial protocol, GCP, and government regulations.
Acknowledgements
Critical Path Institute is supported by the Food and Drug Administration (FDA) of the U.S. Department of Health and Human Services (HHS) and is 55% funded by FDA/HHS, totaling $17,612,250, and 45% funded by non-government sources, totaling $14,203,111. The contents are those of the authors and do not necessarily represent the official views of, nor an endorsement by, FDA/HHS or the U.S. Government. For more information, please visit FDA.gov.
Additional support for the eCOA Consortium comes from membership fees paid by members of the eCOA Consortium (https://c-path.org/programs/ecoac/).
Additional support for the PRO Consortium comes from membership fees paid by members of the PRO Consortium (https://c-path.org/programs/proc/).
Additional Support for the eClinical Forum comes from membership fees paid by members of the eClinical Forum (eCF).
We gratefully acknowledge Andres Escallon and Kelly McQuarrie, BSN, for their inception of this manuscript. The authors wish to thank Susan Britland; Bill Byrom, PhD; Adrian Chan; Celeste A. Elash, MS; Ilan Halberstam; Lobo Loo; Amresh Marunnarkal; Paul O’Donohoe, MSc; Poh Chiam Sim; Aman Thukral, MBA; Donna Wauhop; and Jin Zhou for their contributions towards the development of this manuscript.
Competing Interests
The authors have no competing interests to declare.
Author Contributions
All authors revised the manuscript critically for important intellectual content and approved the final manuscript. At the time this manuscript was being written, Jonathan Gable was an employee of AstraZeneca, At the time this manuscript was being written, Kelly Simpliciano was an employee of Bristol Myers Squibb.
References
1. US Department of Health and Human Services Food and Drug Administration. Food and Drugs, 21 CFR §56 (2021). Available at https://www.ecfr.gov/
2. European Medicines Agency (EMA). Guideline for good clinical practice E6(R2). 2016, p.28, 30. Available at https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-6-r2-guideline-good-clinical-practice-step-5_en.pdf
3. US Department of Health and Human Services Food and Drug Administration. Guidance for Industry: Electronic Source Data in Clinical Investigations. 2013, p.6, 48, Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electronic-source-data-clinical-investigations
4. European Medicines Agency (EMA). Reflection paper on expectations for electronic source data and data transcribed to electronic data collection tools in clinical trials. 2010. p.7, 9, 10 Available at https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/reflection-paper-expectations-electronic-source-data-data-transcribed-electronic-data-collection_en.pdf
5. US Department of Health and Human Services Food and Drug Administration. Guidance for industry: Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. Dec 2009, p.27. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/patient-reported-outcome-measures-use-medical-product-development-support-labeling-claims
6. European Medicines Agency (EMA). Guideline on computerised systems and electronic data in clinical trials Draft. 2021. P.14. Available at https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/draft-guideline-computerised-systems-electronic-data-clinical-trials_en.pdf
7. CDISC Glossary, 2020-12-18. Available at https://evs.nci.nih.gov/ftp1/CDISC/Glossary/CDISC%20Glossary.pdf
8. Center for Drug Evaluation. NMPA Guiding Principles for the Application of Patient-Reported Outcomes in Drug Clinical Development (Trial) Available at https://www.cde.org.cn/main/att/download/35f59af313045f983a8f5bebc23507a8
9. US Department of Health and Human Services Food and Drug Administration, ICH. E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1). 2018 Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r2-good-clinical-practice-integrated-addendum-ich-e6r1
10. Gordon S, Crager J, Howry C, et al, on behalf of the Electronic Patient-Reported (ePRO) Consortium, PRO Consortium. Best Practice Recommendations: User Acceptance Testing for Systems Designed to Collect Clinical Outcome Assessment Data Electronically. Ther Innov Regul Sci. 2022; 56: 442–453. https://link.springer.com/article/10.1007/s43441-021-00363-z. DOI: http://doi.org/10.1007/s43441-021-00363-z
11. Kennedy F, Shearsmith L, Ayres M, et al. Online monitoring of patient self-reported adverse events in early phase clinical trials: Views from patients, clinicians, and trial staff. J Clin Trials. 2021; 18(2): 168–179. DOI: http://doi.org/10.1177/1740774520972125
12. Saeid Shahraz S, Pham T, Gibson M, et al. Does scrolling affect measurement equivalence of electronic patient-reported outcome measures (ePROM)? Results of a quantitative equivalence study. J Patient Rep Outcomes; 2021. DOI: http://doi.org/10.1186/s41687-021-00296-z
13. Ly J, Crescioni M, Eremenco S, et al. on behalf of the ePRO Consortium. Training on the use of technology to collect patient-reported outcome data electronically in clinical trials: best practice recommendations from the ePRO Consortium. Ther Innov Regul Sci. 2019; 53: 431–440. http://journals.sagepub.com/doi/10.1177/2168479018796206. DOI: http://doi.org/10.1177/2168479018796206
14. Fleming S, Barsdorf AI, Howry C, O’Gorman H, Coons SJ. Optimizing electronic capture of clinical outcome assessment data in clinical trials: the case of patient reported endpoints. Ther Innov Regul Sci. 2015; 49: 797–804. https://link.springer.com/article/10.1177/2168479015609102. DOI: http://doi.org/10.1177/2168479015609102
15. Meirte J, Hellemans N, Anthonissen M, et al. Benefits and Disadvantages of Electronic Patient-reported Outcome Measures: Systematic Review. JMIR Perioper Med; 2020. DOI: http://doi.org/10.2196/preprints.15588
16. Aiyegbusi, O. Key methodological considerations for usability testing of electronic patient reported outcome (ePRO) systems. Qual Life Res. 2020; 29(2): 325–333. DOI: http://doi.org/10.1007/s11136-019-02329-z
17. Zbrozek, A, Hebert J, Gogates G, et al. Validation of Electronic Systems to Collect Patient-Reported Outcome (PRO) Data—Recommendations for Clinical Trial Teams: Report of the ISPOR ePRO Systems Validation Good Research Practices Task Force. Value Health. 2013; 16(4): 480–9. ELSEVIER 2013. DOI: http://doi.org/10.1016/j.jval.2013.04.002
Appendix 1. Glossary
| ALCOA+ | Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring and Available, when needed4 |
| Critical data points | Data points determined by the sponsor to be critical to the implementation of the trial, either due to their categorization in endpoint hierarchy of the protocol (primary, secondary, exploratory), or due to their weight in the outcome of the statistical analysis, or due to implications for participant safety. |
| CRO | contract research organization |
| Data originator | Refers to the individual who entered the data in question. This may refer to the participant, the caregiver, the investigator or other site personnel, an observer or other trained specialist with control over creation of source or eSource data. |
| DCR | data change request |
| eCOA | electronic clinical outcome assessment |
| EMA/GCP/IWQ | European Medicines Agency Good Clinical Practice Inspectors Working Group |
| ePRO | electronic patient-reported outcome |
| FDA | Food and Drug Administration |
| GCP | Good Clinical Practice |
| Procedural data points | Data points determined by the sponsor to be non-critical in light of the protocol, the trial objectives, the indication, and the data type. For example, visit label changes may be deemed to be procedural for a trial where timepoint or visit label is collected. This category may also include patient-entered response if it is deemed by the trial team that some patient-entered responses are not critical to the outcomes of the trial. |
| Oversight Plan | A document created in collaboration between the eCOA service provider and the sponsor that explains in detail how data changes will be handled through the life of the trial, including the categorization of data into procedural and critical data points, and the workflow for each. The latter may include sponsor notification to ensure awareness, as well as a request for confirmation from site that the justification for the request is documented. Special instructions, including technical details, where applicable, should be present in this document, as well as any exceptions to the expected process. May also be referred to as “Data Management Plan.” |
| Source | The specific permanent record(s) upon which a user will rely for the reconstruction and evaluation of a clinical investigation. NOTE: The term identifies records planned (designated by the protocol) or referenced as the ones that provide the information underlying the analyses and findings of a clinical investigation. The term is also sometimes used as shorthand for source documents and/or source data. [After ICH E6, CSUICI] See also source document, source data, original data, certified copy.7 |
| eSource | Source record that is electronic. See also source, electronic record.7 |
Appendix 2: Example Oversight Plan for DCR Implementation
| # | Data Change Type | Category | Example | Process followed by eCOA service provider |
|---|---|---|---|---|
| 01 | Demographic changes: YOB |
Procedural or define by sponsor | Site requests updating YOB from 1957 to 1975 to correct user entry error | The eCOA service provider will implement the requested change per the site’s DCR. |
| 02 | Demographic changes: Participant ID | Procedural or define by the sponsor | Site requests updating Participant ID from 1001010 to 1001001 | The eCOA service provider will implement the requested change per the site’s DCR. The eCOA service provider will notify the following key contacts of this request, to check consistency across other databases: |
| 03 | Patient-reported outcome (PRO) measure | Critical | Site requests an update to a response in one of the PRO measures used in this trial: EQ-5D-5L FACT-B EORTC-QLQ-C30 |
eCOA service provider will query back to site for confirmation, using the following language: Thank you for this request. You have requested a change to a participant-entered data point. Please provide the rationale for this change and also confirm that evidence supporting this change is available in the participant’s file. The eCOA service provider will notify the following sponsor contacts when the above query is posted: Sponsor and/or contract research organization (CRO) assess for potential impact to data integrity and participant safety, confirm with site if the data change is supported by source documentation. Sponsor determines adherence to core principles and further actions such as a protocol deviation if necessary. Site confirms, in response to the original query, the presence of adequate “source or supporting documentation” for examples where eCOA is source. The eCOA service provider moves forward with implementing the requested change. |
| 04 | Participant Status | Procedural | Site requests to re-activate a participant who was deactivated in error | The eCOA service provider will implement the requested change per the site’s DCR. |
| 05 | Visit Labels | Procedural | Site requests to relabel Visit X to Visit Y | The eCOA service provider will implement the requested change per the site’s DCR. |
| 06 | Patient-reported outcome (PRO) measure AND Eligibility decision |
Critical | Site requests an update to a response in one of the PRO measures used in this trial: EQ-5D-5L FACT-B EORTC-QLQ-C30 The requested change also impacts whether the participant meets the trial’s inclusion criteria at screening. |
eCOA service provider will query back to site for confirmation, using the following language: Thank you for this request. You have requested a change to a participant-entered data point. Please provide the rationale for this change and also confirm that evidence supporting this change is available in the participant’s file. The eCOA service provider will notify the following sponsor contacts when the above query is posted: In the email to the sponsor, the eCOA service provider will also note that the requested change will result in inclusion of the participant where formerly the participant did not meet criteria for inclusion. The sponsor assesses impact to the participant in the trial; the sponsor confirms with site if the data change is supported with source documentation. Sponsor confirms whether core principles are violated by this request, or whether a protocol deviation is necessary. Site confirms, in response to the original query, the presence of adequate source or supporting documentation supporting the request. Because the requested change may result in an ineligible participant receiving trial drug, the eCOA service provider will ensure that adequate discussion has occurred between sponsor, CRO, and site, before moving forward with the request. |