
1) Learning Objectives
After reading this chapter, the reader should understand:
the regulatory basis for practices in EDC study implementation and start-up
similarities and differences between paper and web-based data collection
basic and common features of fields, forms, and form groupings in EDC systems
common dynamic workflow and data flow options within web-based EDC systems
special considerations for data processing when using web-based EDC
common steps in system set-up and testing
methods for managing system access and privileges
common practices for training clinical investigational sites in the use of EDC
considerations and business models for using vendor-hosted EDC systems
2) Introduction
In the first clinical studies, data were collected on paper forms called Case Report Forms or CRFs. The structured forms served to assure complete and consistent data collection for each study participant. Since the early 1990s, we have documented best practices for designing paper CRFs.1,2,3,4,5 Design considerations focused on graphical layout within the confines of a paper page CRF and visual cues to aid the form filler such as use of boxes versus circles to indicate ‘check all that apply’ versus ‘check one’. Instructions were printed on the forms. There were rules governing the type of writing instruments such as black ballpoint pens. There were rules for correcting data such as use of a single line cross out and providing the corrected value, the date, and the initials of the person making the change. Yet there was nothing to facilitate the workflow of data collection or to prevent writing discrepant or errant values on the paper form.
In the days of paper-based data collection, how the data were entered into electronic format, typically in a Clinical Database Management System (CDMS), was not the most important consideration. Clinical study data were usually double data entered. Entry operators were trained in handling exceptions and allowable corrections. The CDMS partially automated the workflow of data entry, integration of external data, cleaning and coding, and provided automation for tracking data entry, discrepancy identification, and discrepancy resolution. However, the benefits of these advances were largely limited to in-house data management groups in Contract Research Organizations (CRO) or Sponsor organizations.
At the turn of the century, technology leveraging the internet such as web-based EDC systems opened the possibility of extending the benefits of information systems in clinical research to multiple roles on research teams, to investigational sites, and even to study participants. Commercial and academic advances were sporadic.6 EDC systems shifted the work of data entry to clinical investigational sites and contained less data management functionality than the CDMSs of the time. For example, integrating external data and medical coding were challenging or altogether not supported in early EDC systems.
There are four ways through which information systems can add value to organizations: automation, connectivity, decision support, and data mining.7 Examples include (1) automating workflow such as submitting a “low drug supply” alert to the research pharmacy when drug supply is low, (2) providing decision support such as flagging potential toxicities or making real-time status and exception data available to support trial management, (3) providing access to data from other systems through automating pre-population of data from other information systems such as an interface with an eConsent system that registers the participant in the EDC system as enrolled when the eConsent is completed or exchange of participant enrollment data with central or site-based Clinical Trial Management Systems (CTMSs), and (4) data mining through use of study data to identify factors predictive of missed visits or protocol violations. Today’s EDC functionality offers some of this potential. However, achieving broad benefit across clinical study design, conduct, and reporting for clinical investigators, research teams, and study participants is dependent on available EDC functionality and how it is leveraged for a clinical study and how the data are used for better decision-making within and across studies.6,8
At the same time, leveraging advanced functionality lies in the balance between added value and costs. The most recent industry survey reports an average of 68.3 days to build and release a study database, 8.1 days between patient visits and entry of data from the visits into the EDC system, and 36.3 days between last patient last visit and database lock.9 These cycle time durations are longer and more variable than those observed ten years ago.9,10 This chapter focuses on realizing value from EDC functionality through design and implementation of workflow and data flow within clinical studies.
3) Scope
This chapter provides information on the design, development, and implementation concepts related to setting-up a study (sometimes called an application) in an EDC system. Practices, procedures, and recommendations are proposed for clinical data managers to design and implement EDC facilitated workflow and data flow for automation, connectivity, decision support, and data mining within and across clinical studies.
While many of the tasks described in this chapter may be joint responsibilities between different functional areas of an organization, those tasks associated with the collection, processing and storage of data are covered here. These responsibilities are the core of the Clinical Data Management profession. As such, the clinical data manager is usually responsible for the overall implementation of any study application.
Recommendations for EDC system selection were covered in the chapter “Electronic Data Capture – Selecting an EDC System”. Recommendations for study conduct and study closeout using EDC are addressed in the chapter “Electronic Data Capture – Study Conduct, Maintenance and Closeout.”
4) Minimum Standards
As a mode of data collection and management in clinical studies, EDC systems have the potential to impact human subject protection as well as the reliability of trial results. Regulation and guidance are increasingly vocal on the topic. The E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1) contains several passages particularly relevant to use of EDC systems in clinical studies.
Section 2.8 “Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective tasks.”11
Section 2.10, “All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification.”11
Section 5.0 states that “The methods used to assure and control the quality of the trial should be proportionate to the risks inherent in the trial and the importance of the information collected.”11
Section 5.1.1 states that “The sponsor is responsible for implementing and maintaining quality assurance and quality control systems with written SOPs to ensure that trials are conducted and data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirement(s).” Additionally, Section 5.1.3 states that “Quality control should be applied to each stage of data handling to ensure that all data are reliable and have been processed correctly.”11
Section 5.5.1, states that “The sponsor should utilize appropriately qualified individuals to supervise the overall conduct of the trial, to handle the data, to verify the data, to conduct the statistical analyses, and to prepare the trial reports.”11
Section 5.5.3 states that “When using electronic trial data handling and/or remote electronic trial data systems, the sponsor should: a) Ensure and document that the electronic data processing system(s) conforms to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e., validation).”11
Section 5.5.3 addendum states that “The sponsor should base their approach to validation of such systems on a risk assessment that takes into consideration the intended use of the system and the potential of the system to affect human subject protection and reliability of trial results.” and in the addendum b) states the requirement, “Maintains SOPs for using these systems.”11
Section 5.5.3 addendum c-h introductory statement states that “The SOPs should cover system setup, installation, and use. The SOPs should describe system validation and functionality testing, data collection and handling, system maintenance, system security measures, change control, data backup, recovery, contingency planning, and decommissioning.”11
Section 5.5.4 under Trial Management, Data Handling and Recordkeeping, states that “If data are transformed during processing, it should always be possible to compare the original data and observations with the processed data.”11
Similar to ICH E6(R2), Title 21 CFR Part 11 also states requirements for traceability, training, and qualification of personnel, and validation of computer systems used in clinical trials. Requirements in 21 CFR Part 11 Subpart B are stated as controls for closed systems (21 CFR Part 11 Sec. 11.10), controls for open systems (21 CFR Part 11 Sec. 11.30), signature manifestations (21 CFR Part 11 Sec. 11.50), and signature/record linking (21 CFR Part 11 Sec. 11.70). Requirements for electronic signatures are stated in 21 CFR Part 11 Subpart C.12
Recommendations in Section A of the 2007 Guidance for Industry Computerized Systems Used in Clinical Investigations (CSUCI) state that, “Each specific study protocol should identify each step at which a computerized system will be used to create, modify, maintain, archive, retrieve, or transmit source data.”13
Section B of the CSUCI guidance states expectations with respect to Standard Operating Procedures (SOPs): “There should be specific procedures and controls in place when using computerized systems to create, modify, maintain, or transmit electronic records, including when collecting source data at clinical trial sites” and “the SOPs should be made available for use by personnel and for inspection by FDA.”13
Section C reiterates document retention requirements under 21 CFR 312.62, 511.1(b)(7)(ii) and 812.140. Further, section C of CSUCI goes on to state that “When source data are transmitted from one system to another …, or entered directly into a remote computerized system … or an electrocardiogram at the clinical site is transmitted to the sponsor’s computerized system, a copy of the data should be maintained at another location, typically at the clinical site but possibly at some other designated site.” And that “copies should be made contemporaneously with data entry and should be preserved in an appropriate format, such as XML, PDF or paper formats.”13
Section D further specifies 21 CFR Part 11 principles with respect to limiting access to CSUCT (Computer Systems Used in Clinical Trials), audit trails, and date and time stamps.13
Section E likewise provides further detail regarding expectations for security, e.g., “should maintain a cumulative record that indicates, for any point in time, the names of authorized personnel, their titles, and a description of their access privileges” and recommends that, “controls be implemented to prevent, detect, and mitigate effects of computer viruses, worms, or other potentially harmful software code on study data and software.”13
Section F addresses direct entry of data including automation and data standardization; data attribution and traceability including explanation of, “how source data were obtained and managed, and how electronic records were used to capture data”; system documentation that, identifies software and hardware used to, “create, modify, maintain, archive, retrieve, or transmit clinical data”; system controls including storage, back-up and recovery of data; and change control of computerized systems.13
Section G elaborates on training of personnel as stated in 21 CFR 11.10(i) that those who, “develop, maintain, or use computerized systems have the education, training and experience necessary to perform their assigned tasks”, that training be conducted with frequency sufficient to, “ensure familiarity with the computerized system and with any changes to the system during the course of the study” and that, “education, training, and experience be documented.”13
The Medicines & Healthcare products Regulatory Agency (MHRA) ‘GXP’ Data Integrity Guidance and Definitions covers principles of data integrity, establishing data criticality and inherent risk, designing systems and processes to assure data integrity, and it also covers the following topics particularly relevant to EDC:
Similar to ICH E2(R2), MHRA Section 2.6 states, “Users of this guidance need to understand their data processes (as a lifecycle) to identify data with the greatest GXP impact. From that, the identification of the most effective and efficient risk-based control and review of the data can be determined and implemented.”14
Section 6.2, Raw Data states, “Raw data must permit full reconstruction of the activities.”14
Section 6.7 Recording and Collection of Data states that “Organisations should have an appropriate level of process understanding and technical knowledge of systems used for data collection and recording, including their capabilities, limitations, and vulnerabilities,” and that the “selected method [of data collection and recording] should ensure that data of appropriate accuracy, completeness, content and meaning are collected and retained for their intended use.”14
Section 6.9 Data Processing states that “There should be adequate traceability of any user-defined parameters used within data processing activities to the raw data, including attribution to who performed the activity.” And that, “Audit trails and retained records should allow reconstruction of all data processing activities….”14
The General Principles of Software Validation; Final Guidance for Industry and FDA Staff (2002) provides guidance regarding documentation expected of software utilized in a clinical trial.14
Section 2.4 “All production and/or quality system software, even if purchased off-the-shelf, should have documented requirements that fully define its intended use, and information against which testing results and other evidence can be compared, to show that the software is validated for its intended use.”14
Section 4.7 (Software Validation After a Change), “Whenever software is changed, a validation analysis should be conducted not just for validation of the individual change, but also to determine the extent and impact of that change on the entire software system.”14
Section 5.2.2 “Software requirement specifications should identify clearly the potential hazards that can result from a software failure in the system as well as any safety requirements to be implemented in software.”14
Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Volume 4, Annex 11): Computerised Systems (2011) provides the following guidelines when using computerized systems in clinical trials. Though the guidance is in the context of manufacturing, it is included to emphasize the consistency of thinking and guidance relevant to use of computer systems in clinical trials across the regulatory landscape.15
Section 1.0 “Risk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerised system.”15
Section 4.2 states that, “Validation documentation should include change control records (if applicable) and reports on any deviations observed during the validation process.”15
Section 4.5 states that, “The regulated user should take all reasonable steps, to ensure that the system has been developed in accordance with an appropriate quality management system.”15
Section 7.1 states that, “Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability, and accuracy. Access to data should be ensured throughout the retention period.”15
Section 7.2 states that, “Regular back-ups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically.”15
Section 9.0 states that, “Consideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated “audit trail”). For change or deletion of GMP-relevant data the reason should be documented. Audit trails need to be available and convertible to a generally intelligible form and regularly reviewed.”15
Section 10.0 states that, “Any changes to a computerised system including system configurations should only be made in a controlled manner in accordance with a defined procedure.”15
GAMP 5: A Risk-based Approach to Compliant GxP Computerized Systems (2008) suggests scaling activities related to computerized systems with a focus on patient safety, product quality, and data integrity. It provides the following guidelines relevant to GxP regulated computerized systems including systems used to collect and process clinical trial data:
Section 2.1.1 states that “Efforts to ensure fitness for intended use should focus on those aspects that are critical to patient safety, product quality, and data integrity. These critical aspects should be identified, specified, and verified.”16
Section 4.2 states that, “The rigor of traceability activities and the extent of documentation should be based on risk, complexity, and novelty, for example a non-configured product may require traceability only between requirements and testing.”16
Section 4.2 further states that, “The documentation or process used to achieve traceability should be documented and approved during the planning stage and should be an integrated part of the complete life cycle.”16
Section 4.3.4.1 states that, “Change management is a critical activity that is fundamental to maintaining the compliant status of systems and processes. All changes that are proposed during the operational phase of a computerized system, whether related to software (including middleware), hardware, infrastructure, or use of the system, should be subject to a formal change control process (see Appendix 07 for guidance on replacements). This process should ensure that proposed changes are appropriately reviewed to assess impact and risk of implementing the change. The process should ensure that changes are suitably evaluated, authorized, documented, tested, and approved before implementation, and subsequently closed.”16
Section 4.3.6.1 states that, “Processes and procedures should be established to ensure that backup copies of software, records, and data are made, maintained, and retained for a defined period within safe and secure areas.”16
Section 4.3.6.2 states that, “Critical business processes and systems supporting these processes should be identified and the risks to each assessed. Plans should be established and exercised to ensure the timely and effective resumption of these critical business processes and systems.”16
Section 5.3.1.1 states that, “The initial risk assessment should include a decision on whether the system is GxP regulated (i.e., a GxP assessment). If so, the specific regulations should be listed, and to which parts of the system they are applicable. For similar systems, and to avoid unnecessary work, it may be appropriate to base the GxP assessment on the results of a previous assessment, provided the regulated company has an appropriate established procedure.”16
Section 5.3.1.2 states that, “The initial risk assessment should determine the overall impact that the computerized system may have on patient safety, product quality, and data integrity due to its role within the business processes. This should take into account both the complexity of the process, and the complexity, novelty, and use of the system.”16
The FDA guidance, Use of Electronic Health Record (EHR) Data in Clinical Investigations, emphasizes that data sources should be documented, and that source data and documents be retained in compliance with 21 CFR 312.62(c) and 812.140(d).17
Section V.A states that “Sponsors should include in their data management plan a list of EHR systems used by each clinical investigation site in the clinical investigation” and that “Sponsors should document the manufacturer, model number, and version number of the EHR system and whether the EHR system is certified by the Office of the National Coordinator for Health Information Technology (ONC).”17
Section V.I states that “Clinical investigators must retain all paper and electronic source documents (e.g., originals or certified copies) and records as required to be maintained in compliance with 21 CFR 312.62(c) and 812.140(d).”17
Similarly, the FDA’s guidance, Electronic Source Data Used in Clinical Investigations recommends that all data sources at each site be identified.18
Section III.A states that each data element should be associated with an authorized data originator and goes on to state, “A list of all authorized data originators (i.e., persons, systems, devices, and instruments) should be developed and maintained by the sponsor and made available at each clinical site. In the case of electronic, patient-reported outcome measures, the subject (e.g., unique subject identifier) should be listed as the originator.”18
Section III.A.3 elaborates on Title 21 CFR Part 11 and states, “The eCRF should include the capability to record who entered or generated the data [i.e., the originator] and when it was entered or generated.” and “Changes to the data must not obscure the original entry, and must record who made the change, when, and why.”18
Section III.A.5 states that the FDA encourages “the use of electronic prompts, flags, and data quality checks in the eCRF to minimize errors and omissions during data entry.”18
Section III.C states that “The clinical investigator(s) should retain control of the records (i.e., completed and signed eCRF or certified copy of the eCRF).” In other words, eSource data cannot be in sole control of the sponsor.18
As such, we state the following minimum standards for the study implementation and start-up using EDC systems.
5) Best Practices
Best practices were identified by both the review and the writing group. Best practices are not required by regulation or recommended by guidance, but do have supporting evidence either from the literature or consensus of the writing group. As such best practices, like all assertions in GCDMP chapters, have a literature citation where available and are always tagged with a roman numeral indicating the strength of evidence supporting the recommendation. Levels of Evidence are outlined in Table 3.
Minimum Standards.
| 1. | Document requirements for all aspects of the eCRF and data collected, processed, or stored by or in the EDC system. |
| 2. | Document sources of data at each site including explicit statement that the EDC system is used as the source where this is the case. |
| 3. | Ensure data values can be traced from the data origination through all changes and that the audit trail of all data changes is immutable, preserved, and available for review. |
| 4. | Use electronic “prompts, flags, and data quality checks in the eCRF to minimize errors and omissions during data entry.”18 |
| 5. | Establish and follow SOPs to ensure that testing, including user acceptance testing (UAT) of the study-specific EDC application, is commensurate with the assessed risk. |
| 6. | Establish and follow SOPs to ensure that testing is completed and documented prior to implementation and deployment to sites. |
| 7. | Establish and follow SOPs to ensure that all users have documented training prior to using the system. |
| 8. | Establish and follow SOPs to limit data access and permissions to authorized individuals and to document data access and permissions. |
| 9. | Establish and follow SOPs for the process of setting up an EDC system for a study. |
Best Practices.
| 1. | Develop eCRFs with cross-functional teams including but not limited to clinical operations, monitoring, clinical data management, statistics, regulatory affairs, quality assurance, pharmacovigilance/drug safety, and medical leadership. [VI] |
| 2. | Leverage EDC functionality to facilitate the work of sites, monitors, and other study team members to the extent the value outweighs the costs. [III]7,8,19,20 |
| 3. | Design processes to identify and correct data discrepancies at the earliest possible point in study processes. [III]8,20,21,22,23,24 |
| 4. | Ensure adequate attention to the collection, processing, and routing of safety data either through or facilitated by the EDC system. [VI]8 |
| 5. | Warrant the eCRF design is intuitive and user-friendly for sites, monitors, and other study team members and that instructions and references are readily available. [VI] |
| 6. | Help should be available during work days and times of all sites. [VI] |
| 7. | In limited cases the study team may provide data collection forms, instructions and help in local languages. Help should support the number of languages including local dialects needed to communicate with all EDC system users. [VI] |
| 8. | Ensure eCRFs do not introduce bias into the data by containing leading questions or forcing responses. [VI] |
| 9. | Ensure that comprehensive help (written, live, or otherwise) including eCRF entry guidelines, study data definition, and dynamic functionality behavior for all fields, forms, and visits are up-to-date and readily available to sites. [VI]19 |
| 10. | Consider use of available data standards. [VI]8 |
| 11. | Where data standards are used, ensure that the eCRF conforms to the standards so that detail (information content) is not lost in downstream mapping to such standards for submission or data sharing. [VI]8 |
| 12. | The EDC system and all intended data operations such as edit checks and dynamic behavior should be in production prior to enrollment of the first patient. [III]8,9,19,20,21,23 |
Grading Criteria.
| Evidence Level | Evidence Grading Criteria |
|---|---|
| I | Large controlled experiments, meta, or pooled analysis of controlled experiments, regulation or regulatory guidance |
| II | Small controlled experiments with unclear results |
| III | Reviews or synthesis of the empirical literature |
| IV | Observational studies with a comparison group |
| V | Observational studies including demonstration projects and case studies with no control |
| VI | Consensus of the writing group including GCDMP Executive Committee and public comment process |
| VII | Opinion papers |
6) What it Means to Design a Study Application Within an EDC System
Filling in an electronic form is quite different from completing a paper form, where the advantages of EDC technology are leveraged. For example, in an interventional cardiology study, if hemoglobin or hematocrit values below a threshold are entered, a question or form asking about transfusions may be generated as well as assessment information for peri-procedural bleeding. At the same time, an email notification may automatically be sent to the study safety desk. On-screen checks are run to flag out-of-range and logically inconsistent lab values, and the investigator’s assessment of relatedness to the study drug may be required; in this case a discrepant data flag is attached to the form until the investigator’s assessment of relatedness is populated. In this simple scenario, the EDC system added new fields or forms relevant to the patient, provided greater control of data entered in the form, facilitated study workflow, automated tracking of discrepant data, and decreased the gap between the site and central team managing the study – all in real-time. This functionality is not available when collecting data on paper forms and, due to the time-lag in processing and entering paper forms, immediate action by the central study team is not possible. Thus, in this simple example, the EDC system as implemented for the study would provide significant value over paper data collection through automation, connectivity, and decision support.
EDC systems offer the opportunity to define and enforce workflow of data collection in addition to the data to be collected. While the extent to which workflow and data flow can be automated within an EDC system depends on the functionality offered by each system, the basic functionality described in the interventional cardiology study example is available in most EDC software.
It is the increase in connectivity, workflow, and data flow automation and decision support that makes use of EDC different from collecting data on paper forms. An EDC-enabled study goes beyond implementation of new technology. Gaining value from EDC requires re-engineering processes and improving decision-making during study conduct [III].8,19,20,25,26,27 The need for site-facing computer systems to provide benefit to sites in order to garner site support has been articulated since the early days of EDC predecessors.6 The mechanisms of site benefit include the aforementioned ways in which information systems add value to organizations.7 For this reason, setting-up a study in an EDC system is often referred to as “building a study” rather than creating a study database. Using EDC means that for each data value collected on an eCRF, there is an additional choice of what if anything the system should do in response to entry of each of the possible entry values. In today’s EDC systems, there are often many options. Thus, the data manager should have a thorough understanding of workflow and data flow automation and decision support in addition to EDC system functionality to optimize data-related aspects of study conduct. [VI]
How a human works with a paper form and writing instrument is different from working with a computerized system and related input devices. Because EDC is often more invasive than paper data collection in individual working practices and institutional process flow, these interactions become important in design, testing, and implementation. While professionals field-tested paper forms prior to their use on studies, true human centered design, usability testing, or implementation monitoring are often appropriate if not necessary in use of EDC. Further, because EDC systems are used by or touch many roles on study teams, training on use of the system for the study is usually broader than in the past.
7) eCRF Design
Most study builds start with the static aspects of the electronic CRFs (eCRFs), i.e., the data elements or fields to be collected, their definition, valid response values, layout on the screen, and their organization into forms and visits. EDC systems facilitate different ways of grouping data to be collected or displayed on eCRFs. Such groupings include grouping of data collection fields into modules, modules into forms, and forms into visits similar to such groupings on paper data collection forms. (See Figure 1) Likewise, the fields in modules may differ from form to form and the contents of forms may differ from visit to visit. Just as with paper data collection forms, such module-to-module, form-to-form, or visit-to-visit variability increases the development cost and must be weighed against the value added. [VI] In EDC systems, grouping of data elements into modules, forms, or visits may be part of data definition and affect data storage as well as layout. Thus, eCRF design requires a thorough understanding of the relationship between data definition, grouping, layout, and data storage structure in the specific EDC system. [VI]
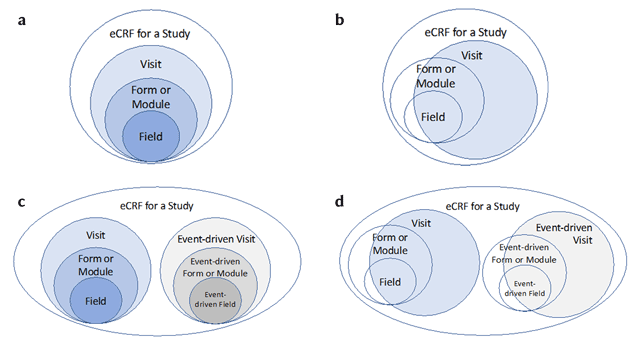
Varieties of Alignment of Fields, Forms, and Visits Commonly Supported by EDC Systems. a: EDC systems often reinforce an alignment between fields, forms (or modules), and visits. Sometimes it is a strict hierarchy. b: Some systems may not require fields, forms, and visits to be associated in a strict hierarchy and may support collection of data outside the study visit structure; i.e., forms that are not associated with visits such as log-based forms that span the study like concomitant medication logs or study withdraw forms. c: Some EDC systems may support event-driven forms and may also have mechanisms or requirements for associating them with visits, other forms, or fields. Examples include repeat assessments or unscheduled visits. d: Some EDC systems may support both event-driven forms and visits as well as forms that occur outside of a visit context. Examples include event-driven forms and log-based forms that span the study such as a log of protocol violations or a log of clinical events.
When designing an eCRF, it is often not known what type of computer(s) will be used for data entry by the end-user. The size and type of screen and input devices such as keyboard and mouse can easily differ across users and at the same time can affect the data entry process. For example, fields “below the scroll” may be more easily missed. Many EDC systems have the capability of allowing for longer or wider forms, as well as multiple ‘forms’ within one eCRF presentation. However, it is good practice to take into consideration the smallest screen available on the market when deciding grouping and lay-out of data fields on eCRF screens even if doing so may reduce the amount of data collected on a single eCRF screen. [VI] Static aspects of eCRF design that are not specific to EDC and are covered in the CRF design chapter of the GCDMP. EDC-specific static aspects of eCRF design are covered in subsequent sections of this chapter.
Most EDC systems have functionality that can be leveraged to guide site staff in data entry and actions to be taken in response to entered data. How the EDC system responds to entered data and other user actions within the system is called dynamic behavior because the system behavior differs based on the user input or action. EDC functionality for dynamic behavior is used to automate or otherwise constrain workflow and data flow in and facilitated by the EDC system. Automation, such as sending a notification email to the safety desk when an adverse event indicated as serious is entered, is a form of dynamic behavior.
Automating workflow and data flow is a major way that EDC systems can provide value to users and organizations. Functionality for automation should be exploited to the extent helpful to users and to the extent practical within a study [VI].7,8 However, workflow and data flow automation requires a significant amount of experience. Design of new automation, i.e., not yet tried by the Data Manager or organization, benefits greatly from application of human-centered design principles such as early and iterative involvement of users in testing dynamic behavior. Automation constrains the options available to system users. Limiting flexibility without careful consideration of all eventual variation that may occur can frustrate users and cause delays when the unanticipated exceptions occur. In a 2003 publication, Kush et al. give an example of an EDC system that automated locking of patient data after internal review such that the sites could no longer make changes. When legitimate changes occurred after internal review, sites were required to call in and request the data to be unlocked. To work around the cumbersome process, sites entered changes into comment fields, which required extensive sorting out prior to analysis.8 Automation and other constraints that affect workflow and data flow, add tremendous value when they work as intended and gracefully handle exceptions. On the other-hand, frequent exceptions or exceptions that are not gracefully handled quickly erode the value gained from the automation. Thus, any automation such as alerts, routing, state changes, or triggering dynamic fields, forms, or visits should be carefully designed and user-tested. [VI]
Automation and other constraints on eCRFs also have the potential to introduce bias into the data by pre-populating or forcing responses. Where such bias is possible, forego the planned automation or constraint. [VI] Statistical review and approval of automation and constraints will help identify potential constraint-caused bias. Data surveillance may be effective in detecting some instances of bias in key safety and efficacy parameters.
Dynamic behavior can be triggered by an individual field or by some relationship between multiple fields or forms. Further, dynamic behavior may act on an individual field or multiple fields. Consider handling of dates. A site in Europe may prefer entering dates using the “dd-mmm-yyyy” format, whereas a site in the United States may prefer using “mmm-dd-yyyy”. Some EDC systems allow site or user-specific settings so that a user can enter dates in their preferred format and the system converts and stores the data in a standard date format. Further dynamic behavior pertaining to dates includes functionality to prevent or facilitate entry of partial dates that may include alerts to the user or flagging values within the database and alternate processing of flagged partial dates. In the date examples, the dynamic behavior is triggered by and acts on single fields (the dates). In this case, the dynamic behavior includes facilitating different entry formats, how the data will be processed by the EDC system, and the workflow and data flow associated with entry of valid values and exceptions. In the interventional cardiology study referenced above, the dynamic behavior includes triggering a new form in response to an entered data value. The example illustrates dynamic behavior triggered by indication of a transfusion (an individual field) and acting on multiple fields through creation of a new bleed form with fields for bleed-related lab values and bleed assessment details. Thus, when designing an eCRF, the designer evaluates each field for whether it is a trigger for dynamic behavior either alone or in concert with other fields and whether the desired behavior pertains to an individual field or to multiple fields. [VI]
Dynamic behavior can be confusing to system users because by definition the system behavior differs based on the input. Thus, dynamic behavior should be documented and emphasized in training [VI].19 Dynamic aspects of EDC study builds are covered in subsequent sections of this chapter.
Dynamic behavior may also be used to facilitate study operations such as Source Document Verification (SDV). For example, in 2008 Nahm et al. recommend leveraging the EDC system to support field-level SDV to support point estimation of the source-to-EDC error rate [III].28 A similar process has been recommended for measuring the medical record abstraction error rate.29,30 Other operational processes such as two-rater classification of clinical events and adjudication can also be facilitated through dynamic behavior in EDC systems. Use of EDC functionality to facilitate study operations should be used where the value added outweighs the costs. [VI]
Whether static or dynamic forms are used, use of data standards and form libraries to facilitate reuse of eCRF forms and their features can decrease costs associated with study start-up [VI].8,20
8) Basic Form Features
The most basic function of EDC software is the ability to build and deploy web-based electronic forms for the entry of data and to store the entered data. In most EDC systems, data elements are associated with a data collection structure when they are first added to the system. At this time consideration should be given for use of special characters that could potentially be in use. [IV.] Every time the data element is implemented as a field in a form, the associated data collection structure is used, standardizing it throughout the study. Common data collection structures in EDC systems include free text, many options for semi-structured text, radio buttons, dropdown lists, and checklists. The use of pre-defined answer choices such as those in radio buttons, checklists, and dropdown lists provides constraints during entry and, along with on-screen edit checks, are associated with higher data quality.31
Free text fields allow the user to type in character strings. Free text fields often require specification of a length where the length is sometimes limited by the system or by functionality in downstream data systems. Consistency of responses is challenging with completing free text fields and, for this reason, with the exception of verbatim text to be coded later, they are rarely used for safety and efficacy endpoints. Free text fields are used when constraining the possible responses is not desired, for example, comment fields or collection of site explanations of protocol deviations.
Semi-structured text fields, however, are used often and have many variations. For example, response characters can be limited to alpha or numeric characters. Integer data can be collected and limited to a number of integers. Floating point, i.e., numbers without a fixed number of digits before or after the decimal, and fixed point, i.e., numbers with a fixed number of digits after the decimal, can be used where fractional parts are expected. Semi-structured fields may also constrain the format of entered data, such as parentheses around a phone number area code and a dash between the third and fourth digit or specification of a date format. Semi-structured text fields constrain entered data and increase consistency in the collected data. Semi-structured fields should maximally constrain entered data while accommodating entry of all possible accurate response options. [VI] For example, assuming the system has the ability, the numeric data element heart rate for adult humans in beats per minute should be constrained to an integer value. While there will be disagreement about the range of values possible, the fastest reported human heart rate is 480 beats per minute.32 A range of between zero and 250 beats per minute would not be unreasonable for a field constraint for a study in normal human adults. The aforementioned recommendation requires balancing clinical representativeness for the rare case such as 480 bpm or zero at death with the error-prevention benefit of a tighter range. Consideration should also be given to fields of mixed type. For example, many lab values are typically a numeric field, but some test results may be reported with a >, <, or + symbol, or as ‘positive’, ‘negative’, ‘few’ or ‘trace’.
Radio buttons allow selection of only one choice from a usually short list of options. Radio buttons provide the maximum constraint possible. As such, this data collection structure should be used when the response options are known and standardized. [VI] Implementation of radio buttons should include a mechanism to de-select, i.e., un-select, a previously selected response. [VI]
Like radio buttons, checklists are usually used for shorter lists because all response options are displayed on the screen. Checklists differ from radio buttons in that they allow the user to select more than one option, i.e., check all that apply. Also, like radio buttons, checklists should be used when the response options are known and standardized. [VI] Implementation of checklists should include a mechanism to de-select, i.e., un-select, a previously selected response. [VI]
Dropdown lists allow the selection usually of only one choice and provide a significant amount of constraint. A drop-down list can be used in any situation appropriate for radio buttons but require one or more additional clicks; thus, for short lists, radio buttons are preferred. [VI] Dropdown lists are often used for longer lists to save space on the screen. However, implementing dropdown lists where a scroll is required should be approached with caution because items “below the scroll” may be more likely to be missed. [VI] A variation on both the free text and dropdown list is type ahead functionality where the choice options are restricted by matching the options to the characters typed by the user. Type-ahead functionality in combination with a dropdown list may allow use of very long lists including some clinical controlled terminology sets such as the International Classification of Diseases, or Current Procedural Terminology. Implementation of checklists should include a mechanism to de-select, i.e., un-select a previously selected response. [VI]
Radio buttons, checklists, and dropdown lists collect discrete response options. The underlying data, however, may not always be discrete. For example, an eCRF may collect the following data element, “Does the participant have hypercholesterolemia?” with the response options yes and no, rather than collect the raw cholesterol value. This discretization also represents a clinical diagnosis that admittedly may take into account more than a single lab value, the discretization reduces the information content of the data. The yes/no response would be useless if a different definitional range for hypercholesterolemia were to be applied. Continuous ratio or interval data should be discretized to ordinal or nominal data only after careful consideration. [VI]
9) Required verses not required fields
A field stated as required in a study protocol may or may not be implemented as such in an EDC system. Certain data may be critical to safety or efficacy endpoints of a study. However, there may be times when the data are legitimately not available. Functionality for implementing a field as required in an EDC system differs across systems. In some systems, marking a field as required means that the user cannot save the form or move past the field or form until a value is supplied. This is often called a “hard stop” or a “hard required”. Many EDC systems offer an override feature where a user is prompted to provide a missing value and allowed to still move forward without one. This is sometimes called a “soft required” meaning that a value for the field is expected and if the needed values are not entered, the user can acknowledge or override the alert. The missing value may or may not be tracked depending on the functionality offered by the EDC system. Whereas paper-based data collection mandated a query be sent to the site after missing data were discovered, in EDC this can be a one-step process, with the user confirming that the value is missing and, possibly, providing a reason why. Where data is expected to be missing, it may be appropriate to include response options for the user to indicate a reason the data are not provided, e.g., “sample not collected”, “assessment not done”, “not applicable”, “data not available/not retrievable”, “asked but unknown”, “asked but subject refused to answer”, “actual value invalid”, etc. [VI]
Some data are conditionally required. For example, a pregnancy test result is often conditionally required based on gender, and the specifics of an adverse event are required when an adverse event is indicated as having occurred. Many EDC systems include functionality to implement hard or soft constraints for conditionally required data. Implementing such functionality decreases user data entry time and frustration and should be implemented where it exists. [VI]
10) Calculated Derived fields
Most EDC systems have the ability to derive fields using basic calculations and algorithms. For example, Body Mass Index (BMI) is a calculation dependent on the subjects’ height and weight. A field may be placed in the eCRF that automatically calculates and displays the BMI during entry for the site. Other examples include unit conversions, calculating weight-based drug dosing, scoring rating scales, and applying eligibility and other study criteria to raw data. This functionality is useful in providing decision support to sites. Similar to calculated fields, there are times when it is helpful for a site to have the ability to ‘see’ the data that was entered at a previous study time point, form, or visit. The user should not be able to change the re-displayed data copied into the current visit. [VI] If the original data changes, the calculated or copied value should automatically be updated. [VI] How copied or calculated fields work should be emphasized in training and a mechanism should be in place to indicate to the user how and why data has appeared in a form. [VI]
Algorithms to calculate values may be run at the time of entry or afterward and stored but not displayed. Algorithms to calculate values should be run at the time of entry and calculated values should be displayed if they are used in decision-making at the clinical investigational site or if edit checks are based on the calculated value. [VI] If calculated derived fields are to be used, they should not be editable by the site, to ensure a consistent calculation is performed across the study population. [VI] Calculated derived values for the purpose of data analysis are usually not programmed into EDC systems; rather, they are programmed as part of building analysis data sets. Calculations are the result of algorithms; like other computer programs, procedures should exist to determine the extent of testing and the processes by which testing and documentation of testing should occur [I].12
11) Dynamic fields
Many EDC systems have the ability to conditionally display a field (or not), for example, based on a previously entered data value. In this case, the field on which the condition depends is referred to as a trigger field, and the conditional logic is referred to as the trigger. This is often referred to as ‘skip logic’, ‘skip pattern’, ‘dynamic branching’, or ‘dynamic field’. For example, if collecting whether a procedure was performed, a lead-in question might be asked, “Did the subject complete the procedure?” If the answer is “Yes”, then the eCRF will display questions specific to the procedure. If the answer is “No”, then the eCRF will show only a drop-down select list or text box for the site to record the reason the procedure was not performed. Displaying or activating fields only when a response is valid is a form of constraint and prevents discrepant data from being entered. Such constraints should be implemented where feasible. [VI]
Some systems perform this feature in real time while other systems apply such rules once the form is saved. In the latter case, the functionality is limited to fields on subsequent forms. If the EDC system supports complex dynamic field branching in ‘real time’, sometimes called multi-layered dynamics, then use of the feature to control entry is recommended to catch errors at the earliest possible point or to altogether prevent them. [VI] If the EDC system does not have real-time branching functionality, clear instructions for completion as well as edit checks to catch logically discrepant data should be used. [VI] Dynamic field behavior should be emphasized in training and a mechanism should be in place to indicate to the user how, why, and when a dynamic field appears. [VI]
12) Dynamic Forms
In EDC systems, fields are associated with forms (or item groups or modules of forms), and forms may (Figure 1a) or may not (Figure 1b) be associated with a study visit. Similar to dynamic fields, many EDC systems support dynamic forms, i.e., forms that appear only when a subject meets a certain criterion such as entry of a particular data value. A common example of a dynamic form is a form for prostate cancer screening that only needs to be completed for male participants. Because dynamic forms do not always appear to be available in the EDC system, e.g., the prostate screening form will not appear in the system if a participant is female, they have the potential to confuse users. Dynamic form behavior should be emphasized in training and a mechanism should be in place to indicate to the user how, why, and when a dynamic form appears. [VI]
Use of dynamic forms requires considering how the data on dynamic forms activated in error are handled when the form is subsequently inactivated. For example, consider the case where the sex of a patient is incorrectly entered as female, generating dynamic gender-specific forms; afterwards, the gender is subsequently corrected to male. Different EDC systems handle this scenario differently. Because the origin and all changes to data should be recorded and immutable,11,12 the removal of dynamic forms generated in error and any data entered on them should be permanently tracked by the system. [I]
Dynamic forms such as repeat, event-driven, or unscheduled assessments can be automatically triggered as just described or they can be manually triggered. Some EDC systems support repeat form functionality where a form can be set up to allow site users to manually trigger a new instance of the form. For example, some studies may allow for or require repeat assessments for abnormal vital sign, laboratory, or ECG results. These could be implemented as a repeat form if the EDC system supports this functionality. Using built-in system functionality for repeat forms often also automatically maintains the association of the records for repeated assessment results with the visit in which the original result was measured. However, there is substantial variability in if and how EDC systems support repeatable forms; for example, some systems only allow for repeatable forms in association with unscheduled visits. Manually triggered dynamic forms such as repeatable forms are usually used when the necessity of the additional form instance is (1) dependent on the participant’s course or resulting data or (2) only occurs for a subset of the participants.
These manually or “site user-triggered” forms require consideration at set-up for how to maintain the association of the data with the appropriate time point or visit. In other words, where the EDC system does not or cannot support automatic association of dynamic form data with the needed time point or triggering event, special provisions for referential integrity must be made, such as requiring entry of the number or date of the event or visit with which the data on the new form should be associated. [VI] Further, the appearance of the new instance on the system should be clearly distinct from a visit. [VI]
Many EDC systems have built in functionality for the user to enter the actual date of data collection and associate the date with the data collected. This data is both ‘meta-data’ (data about the data), as well as part of the clinical data in the EDC system. It is often assumed that an assessment or collection date can be derived from the visit date if needed. Thus, physical exam or vital signs forms often lack assessment or collection date, presumably to decrease data entry burden on the site. However, some studies permit assessments to occur within a window of the scheduled or actual visit. These are often implemented as dynamic forms. Omitting a visit date for dynamic forms or forms within the visit results in information loss by losing the association of the assessment data with the actual date on which it was collected. We recommend explicit association of the data to the collection date for better traceability as articulated in ICH E6(R2). [VI]
Determining how to best implement dynamic forms depends on capabilities of the EDC system and complexity of the study. When used multiple times within a study or program, how dynamic forms are triggered and completed and how the content and behavior appear to site users should be consistent to decrease the potential for user confusion. [VI] For example, consistently using dynamic forms in similar situations such as all repeatable forms or event-driven forms, and using the same process for triggering, completing, and correcting them across all form instances will go a long way toward usability.
13) Dynamic Visits
Similar to forms, some visits are expected for a study, i.e., scheduled per protocol, whereas others are conditional, that is some visits are only needed for a subset of the participants and are driven by events that occur during the study. Expected visits are usually described as such in the study visit schedule within the protocol and implemented as such in the eCRF design and EDC system. Like event-driven forms, event-driven visits may or may not occur and are created as needed. Event-driven or otherwise unscheduled visits are not expected to occur for every participant. Many EDC systems support event-driven visits by facilitating their display (or not) based on data entered for a patient or by manual triggering. In an oncology trial for example, when a patient meets a certain criterion he or she may move to a different treatment group with a different set of visits. In some EDC systems, the ability to auto-skip or hide visits is an option. For example, a field is answered in a visit form stating the subject discontinued from the study and the remaining visits are skipped or hidden from the visit schedule and are no longer expected for that subject.
Similar to their form counterparts, referential integrity for repeatable (sometimes called multi-occurring) and event-driven visits often requires special consideration. For example, expected visits have a minimum count of one and a maximum count of one while event-driven visits may or may not occur and they may occur multiple times for a patient within a study; i.e., a minimum count of 0 and often a maximum count greater than one. Providing for referential integrity means that the data collected on dynamic visits should be associated with the correct triggering event as well as the correct time point. To accomplish this, typically these visits should be declared and set-up as such, as noted in Figure 1c and 1d. Dynamics may impact data entry efficiency and system speed so clinical data managers should weigh the benefit versus the possibility of overloading sites with confusing or complicated dynamic functionality when considering dynamic visits. [VI] Like dynamic forms, dynamic visits should be taken into consideration in data status reporting.
14) Decision Support
Decision support is one of the major ways that information systems can add value.7 Automation, discussed previously, is one way of providing decision support. Automatically sending an email notification to the medical monitor when an adverse event indicated as serious is reported is an example of automation. Decision support may leverage automation; for example, the alert to the medical monitor might include additional data and highlight fields pertinent to decision making. Signal detection algorithms that run over data as they are entered or run nightly to detect visits outside the protocol-defined window, disruptions in study medication adherence, or prohibited concomitant medications that trigger automated alerts to the site and site monitor are other examples. Collecting raw data and having the EDC system provide the calculated values in real-time when sites require the calculated values for decision-making and are using the EDC system when making the decision, such as having the EDC system calculate weight-based heparin dosing for a trial in the operative setting is another example. Not all decision support leverages automation. Data status reports and exception reports, discussed in EDC Chapter 3 “Electronic Data Capture – Study Conduct, Maintenance, and Closeout” are examples of decision support without automation as are data visualizations to support signal detection. To be effective, the decision support must be provided at the time and location where the decision is being made. Additional examples of decision support in conjunction with automation provided to study participants and study personnel are described in Cramon et al. (2014) and Mitchel (2008).20,33
15) Form Instructions
From a cognitive engineering standpoint, forms serve as an extension of an individual’s thinking.34 Good form design minimizes cognitive load on the user, i.e., the number or complexity of mental operations that a form completer needs to perform. Thus, the best form completion instructions are those that are not needed because the structure of the form makes correct completion obvious and prevents incorrect completion. In the remote collection of study data, as in the case with EDC, such extensive constraint is often not possible. For example, where a site has only year or month and year for a medical history item, studies would rather have lower resolution than no data. Sometimes the flexibility is needed to account for expected variability while in other situations the lack of system functionality requires it. Form completion instructions fill the gap between the ideal of complete constraint and the reality of needing some flexibility on the user interface and are usually required. Most EDC systems offer options for form completion instructions that go far beyond those available to studies using paper forms. For example, most EDC systems provide additional opportunities to co-locate instructions with the fields to which they pertain via mouse-over or other field-specific and user-activated help. Field-specific instructions should be as close as possible to the field to which they pertain and available to the user with minimal barriers to access.35 While this leans away from provision of form completion instructions as a separate document, instructions are the minimum requirements to support consistency where options exist. [VI] A separate instruction document is better than optionality without clarification. [VI] There is a trade-off between (1) the need to cover options with instructions and (2) the amount of time needed to specify and add them into an EDC system. For more information on form completion guidelines, refer to the GCDMP’s CRF Design and CRF Completion Guideline chapters.
16) Data Integration Set-up
Data independence is the ability to change data values and logical or physical structure of the data without changing the software application that uses the data.36 Most clinical research software today utilizes an independent underlying database management system. Thus, referring to the “clinical database” today usually means the data stored in the database utilized by the EDC software.
The capability to extract data from database systems and transfer the data to another system is a feature of all modern database systems. Yet integrating data from other sources with EDC systems remains a challenge for 77% of respondents to a recent industry survey.9,10 The major areas of difficulty included (1) integration issues, (2) EDC system limitations, and (3) technical demands of support staff.9 At the same time, independent surveys have documented the increase in number of data sources in clinical studies9,37,38 and a doubling of the number of data points collected for protocols between 2007 and 2017.9 The two most recent surveys reported that 100% of respondents’ studies use EDC.9,38 Thus, it is not likely that EDC will be eclipsed in the near future. However, the proportion of study data collected through EDC systems has begun to give way to the increasing volume of data collected through other data sources.10 For these reasons, integration of data from other sources with data collected through EDC systems grows in importance. Though not always the case, assuming that all collected data will be analyzed implies that the data will be integrated at some point between their acquisition and analysis. The best practices of (1) active management through near real-time data acquisition and review (see EDC Chapter 3), and (2) identifying and resolving data discrepancies and operational problems at the earliest practical point in time [III]8,20,21,22,23,24 give heavy weight toward integrating data earlier rather than later in study data processing pipelines.
The functionality needed for data integration and associated data processing should be decided and planned early and set-up along with the study EDC system. [VI] General methods for integration of external data are described in the GCDMP chapter on Integration of External Data. There are multiple approaches to integrating data on studies using EDC for clinical data capture including: a) importing batch data in the EDC system, b) integrating data in a separate repository, c) real-time or near real-time interfaces between EDC system and other systems and d) relying on sites accessing an external system for during-study data needs and integrating the data after the fact. These main approaches are described below.
a) Importing batch data in the EDC system
Most EDC systems offer functionality to import, integrate, process, and display externally collected or externally managed study data such as data from central clinical labs, core labs, ePRO systems, and central reading centers. Some EDC systems support only batch data transfers in favor of live system-to-system interfaces. EDC systems offer different levels of functionality for common processing of imported data such as staging incoming data files, pre-load exception checks, importing the data into the EDC system, edit checks to reconcile imported data with other clinical data, and functionality to track the disposition of identified discrepancies.
Though vendors have argued for the EDC system to serve as the data integration hub for studies and organizations,26,39 such comprehensive integration is seldom the case. Some EDC systems require manual imports of external data. Support for cumulative versus incremental imports varies. Some EDC systems do not support standard data processing functions such as discrepancy identification, discrepancy resolution, and change tracking for imported data. The decision to import and integrate external data depends on the study needs, the functionality available in the EDC system, and the resources required to apply that functionality. Clinical data managers should understand how data collected or maintained outside an EDC system will be used, who will use it, and for what purpose. Into which system changes to external data will be made, which system will maintain the audit trail, and how the audit trail information will be exchanged are just as important. The answers to these questions help determine the extent and timing of data integration. Similar situations exist with exchange of data between EDC systems and common clinical study infrastructure systems such as pharmacy, safety (where safety data are managed in a separate system), and CTMS systems. Thus, the best practice recommended here is to integrate data where the data are needed for site-decision-making and the value of doing so outweighs the costs. [VI]
b) Integration of data in a separate repository
Integration of EDC data with data from other sources in a separate repository outside the EDC system treats EDC as just another data source. Some systems support a repository or warehouse associated with but outside the EDC system for this purpose. Others mention use of a CDMS39 or other products.40 While there are not industry accounts of these in the published literature, they are likely common within organizations. Where data from the other sources are not needed by the sites for decision-making, integration in a repository or study data warehouse may be an acceptable and efficient solution.
c) Integration of EDC systems with pertinent other data sources
Some EDC systems and some external data providers support direct interfaces for real-time or near real-time data exchange. Historically these have taken more effort to set up than using existing EDC system functionality for batch imports. However, standards and technology to support interfaces and configure them within reasonable timeframes are now available. Haak reports such integration with imaging.41 Franklin described a similar approach of setting up point-to-point interfaces with other systems as needed at a large academic institution.42 Lu provides an example of multiple such interfaces with a commercial EDC system to support post-marketing studies.27 Today, direct interfaces are likely more common with infrastructure systems such as drug-supply management, financial site payment systems, and enterprise Sponsor or CRO Clinical Trial Management Systems (CTMSs).
d) External site access to external data
Some vendors involved in the collection and management of data from central clinical labs, core labs, ePRO systems, and central reading centers offer real-time access to an information system where the external data can be viewed by sites. However, these often require a separate login. Integration of external data into an EDC system may be required if the data have direct impact on clinical decisions or study management. Examples include where EDC-based randomization uses scores on patient-completed assessments or when doses are adjusted based on results from an external lab. When data are needed and expected to be used by site users of the EDC system, they should instead be integrated into the EDC system and the value of doing so outweighs the costs. [VI]
Because integration of external data usually includes reconciliation and cleaning of the data, integration of external data into the EDC system also facilitates interim analysis and database lock by these checks having been conducted in an ongoing manner throughout the study. Where it obviates the need for manual entry of data, integration of external data likely saves time and increases data quality. Maintaining a study blind is an additional consideration in integration of external data. Data with the potential to unblind a blinded study, for example, a lab result that might give away the treatment assignment, may require a separate integration strategy to both accomplish ongoing reconciliation and at the same time maintain the blind.
Setting up an EDC system to receive imported data usually requires creating data fields within EDC system to receive and store the data to be integrated as well as the algorithms through which the incoming data are parsed, transformed if necessary, and written to the destination fields. Because data integration requires algorithmic or manual manipulation of data, the planned data integration should be fully specified, tested, and traceable [I].12 Detailed considerations and practices for designing, specifying, managing, and assuring the quality and compliance of externally managed data can be found in the GCDMP chapter titled Integration of External Data.
17) Data Validation Checks (Edit Checks)
Data validation checks are algorithms that are used to screen data for invalid, questionable, or anomalous values. They are sometimes referred to as edit checks, query rules, or error checks. Data validation checks that identify problems as data are entered in EDC systems are also referred to as on-screen checks. EDC systems vary widely in the workflow related to query processing and status, for example whether on-screen checks that fire before data are committed to the database are tracked as discrepancies. A thorough understanding of the functionality and associated metadata is required to optimize processes using on-screen checks. Edit checks should be developed concurrently and iteratively as part of the eCRF with the eCRF specifications finalized prior to the edit check specifications. [VI]
On-screen checks enable enterers to address the flagged values sooner if not immediately, ideally during the assessment or when the source of the information is at hand. Preventing errors or catching data problems earlier reduces costs. It is widely accepted that there are significant increases in total cost the further downstream errors are caught. This concept has become known as the 1-10-100 rule and is described in three stages, as error prevention is ten-fold less than correction where correction is yet again ten-fold less expensive than remediation of failures due to uncorrected errors.43 Thus, in a risk-based approach, costs associated with prevention can be weighed against cost of correction and damages from failures due to uncorrected errors. Further, use of on-screen edit checks with single-entered data is associated with data quality similar to that of double entered data.44 On-screen checks should be used with EDC to the extent that benefit outweighs cost associated with, for example, human safety, re-work, and regulatory delays [III].44
Operationally, on-screen checks in EDC systems increase the immediacy with which Data Managers, study Monitors, or in-house Study Coordinators or Site Managers can become aware of and review unresolved discrepancies and interact with investigational sites to resolve them. Such data-driven contact by phone with site staff promotes an active approach to decreasing elapsed time to complete and clean data. See EDC Chapter 3 for more information on active study management. Getting data in and clean faster has always been a major part of the value proposition of EDC. At the same time, because EDC broadened the number and variability in users from internal personnel to users at the clinical sites involved in a study, the requirements for system training and usability are significantly increased.
a) Types of Edit Checks
Most EDC systems use a rules-based approach to identification of discrepant data and have functionality for authoring, storing, managing, executing the rules and tracking the lifecycle of identified discrepancies. Edit checks in EDC can be classified into two broad categories, “hard” edits and “soft” edits. Soft edits identify discrepant data and usually prompt the site for data correction but allow the data to be confirmed as is and saved so that entry can continue. Whereas hard edit checks also identify discrepant data but prevent the identified data from being saved. In some systems, the form itself cannot be saved with open hard edits. In other systems, hard edits do not produce an alert that a user can “confirm as is” or override. Thus, hard edits are sometimes called non-actionable because the user cannot acknowledge the check and proceed; the only permissible action is to enter data that conform to the requirements. Data type checks (sometimes called browser checks because they almost always run real-time in the browser) are commonly implemented as hard edits. For example, if a user attempts to enter an alphabetical character in a numerical field, the check will not accept the data and if the field is required, the form will not save until conformant data are entered. Another type of a hard edit is a property check. Property checks prevent entering data that do not match form and/or item property settings of the field documented during system set-up. For example, when a field requires a number with 2 decimals, a value of “3” cannot be entered. Instead, a number with two digits to the right of the decimal must be entered to satisfy the property requirement. Without satisfying the property requirement, if the field is required, the form will not save until conformant data are entered. For this reason, hard edits are usually used for non-feasible scenarios such as physically impossible values while soft edits are used to identify data values that are unexpected or unlikely but which could occur. These considerations are more important in the context of EDC because the data enterer is at a clinical investigational site. Because failure of a hard edit prevents forward progress with the task of data entry, users are incentivized to enter a data value that will “pass the check”. Thus, we do not recommend use of hard edits in EDC. [VI] Many systems have evolved and now allow all checks to be implemented as soft edits and allow entry of otherwise invalid data along with a reason for the non-conformant data.
b) Lifecycle Documentation and Management of Edit Checks
Because data entry is done by investigational sites with EDC, the user interface and usability become more important. If a discrepant data value is identified by an edit check, a real-time indicator such as a color change, an audible alert, haptic feedback, or a change in iconography on or near the discrepant data is most helpful to the user. [VI] Similarly, an explanation of the discrepancy should be readily available, whether a single data point error or an erroneously fired query that generates multiple errors. [VI] In addition to real-time cues to the user, a lifecycle record for all detected discrepancies best meets the traceability requirements as stated in ICH E6(R2) [I].11 Such a record provides a mechanism through which changes to data can be reconstructed from the original entry, a prompt (or not) regarding a discrepancy, and changes to data. In the absence of such a record, it is not possible to distinguish prompted versus unprompted changes to data following the initial entry. Further, a record of open discrepancies facilitates reporting and active management of data collection and cleaning. EDC system functionality for lifecycle documentation and management of discrepancies varies.
Usability and lifecycle documentation and management of data discrepancies can be disrupted where EDC functionality does not support complex multivariate checks. For example, complex rules such as those that cross multiple forms or visits or those with logic requiring extensive programming may not be able to be implemented real-time on the user interface or, in some cases, implemented at all within the EDC system. EDC systems vary and a check may be considered complex by one system and easy for another. Where such complex checks are considered required for the study and cannot be implemented within the EDC system, they must be developed and implemented outside the EDC system. Consequences of developing and implementing edit checks externally include inability to execute them in a real-time manner on the user interface, cost and time to maintain the external systems, additional resources required to track and report externally identified discrepancies, and challenges providing comprehensive status and work-facilitating reporting during the study. For example, how will the check results be communicated to the users via the EDC user interface if they are not themselves implemented within the EDC system? Without an interface, manual re-entry of queries into the EDC system is often performed so that site-based users can use the EDC system to resolve queries. The resources needed to manage this activity should be considered. These realities erode the benefit of EDC.
In addition to edit check complexity, other factors such as data availability and system performance may prompt consideration of implementation of edit checks external to the EDC system. For example, if an edit check uses coded terms but coding occurs external to the EDC system, unless data are also coded automatically or by the site users within the EDC system, the edit check cannot run in real-time. Further, for edit checks to be run and tracked within the EDC system the coded data must be imported into the EDC system or be available to the EDC system through an interface. A second factor that commonly prompts implementation of edit checks outside the EDC system is system performance in the presence of numerous checks or complex checks programmed in EDC. For example, suppose there is a need to check that the last date of subject contact is the last chronological date in the database. In this case, the edit check should pull all dates from each module in the database and compare those dates against the newly entered date of last contact, or prepopulate it directly. This type of edit check might access the underlying database thousands of times and noticeably degrade system response time. Optimization of system performance may require balancing running complex checks real-time, system response time, and infrastructure cost and most often requires collaboration with Information Technology professionals because of the interplay between hardware and software or optimization of computer programs that run on the underlying database. [VI] The trade-offs between real-time identification and resolution of discrepancies and data availability and system performance may also erode the benefit of EDC.
Usability and lifecycle documentation and management of data discrepancies can be disrupted where EDC functionality does not support graceful lifecycle management of the edit check rules themselves. To prevent frustrating and time-consuming rework from data discrepancies identified in data that previously appeared to sites as complete or clean, edit checks should be available when the system is moved to production. [VI] While this recommendation is straightforward when starting a study, mid-study changes bring challenges and trade-offs. EDC systems have different limitations when adding or altering edit checks after data have already been entered. For example, while some systems have the ability to re-trigger edits on existing data when edit checks are added mid-study, other systems may only apply new edit checks to new or modified data. Therefore, the clinical data manager should consider how existing data will be checked and may need to provide for checking previously entered data by means such as programming a listing to identify issues with existing data. In this example, sites should also be informed that they may be required to resolve issues identified in earlier visits.
Similarly, usability and lifecycle documentation and management of data discrepancies can be disrupted where EDC functionality does not support control over when checks are run. Edit checks may generate queries due to the order of data entry. For example, consider an edit check that compares the date for visit 3 to the date from visit 2 to assure that visit 3 occurs after visit 2 in time. If data for Visit 3 is entered prior to visit 2, i.e., out of the expected sequence, the check triggered from entry of the visit 3 date may not run on entry without the comparator record for visit 2 and may not run when visit 2 is subsequently entered because the check is triggered from visit 3. These scenarios depend on the functionality supported by the EDC system. Some systems may allow manual re-execution of all checks to ameliorate this problem whereas others may not.
EDC system functionality for developing and managing edit checks varies. For example, some systems handle univariate checks in the data element or screen definition process. Univariate checks, those that apply to a single data value, specify valid values of a data element and thus are sometimes viewed as properties of the data element. Examples of univariate checks include data-type checks, missing checks, value options for enumerated data elements, and maximum, minimum, or range checks for numerical data elements. Univariate checks are often specified and managed as part of the data element definition process within EDC systems. The resulting data definition is then leveraged to offer automated checking of entered data against the valid value constraints in the data definition. The type and extent of definitional information entered and the extent to which EDC systems leverage it for checking data vary across systems. Importantly, edit checks that rely on properties or similar metadata do not require computer programming and thus data definition-based checks are not subject to the requirements of software validation; i.e., they do not need to be tested for each study set-up within an EDC system once the relevant functionality is validated. Not all univariate checks can be supported by definitional metadata, for example conditional univariate checks such as those that apply in some situations but not in others. Univariate checks that have to be programmed as edit checks must be tested just like any other computer program [I].11
Multivariate check functionality varies even more. Recall that multivariate checks are those that compare multiple data values, for example, comparing subject weights over time use unlikely weight changes to identify potentially errant data. These checks usually require writing rules (logic-based algorithms). EDC systems vary in the extent of support for authoring and managing such rules with some systems merely storing executable SQL code written for the system’s data model. Such rules are custom computer programs and should be tested as such [I].11 Guidance on rules-based approaches to data cleaning and methods for developing, testing and managing rules can be found in the GCDMP chapter titled, “Edit Check Design Principles”.
To define and review edit checks prior to production release of an EDC study, clinical data managers may coordinate activities with clinical, IT, quality control, quality assurance, programming, statistics, or other groups. Because of the aforementioned trade-offs and impact on study operations at sites, the approach to data cleaning should be discussed during development of the EDC study specification, and in consultation with all stakeholders involved in data validation, especially sites and team members who work directly with sites [VI].
18) Medical Coding Set-up
During the eCRF development process, all data fields to be coded and the controlled terminologies with which they will be coded should be identified. [VI] Decisions about coding of medications, concomitant conditions, adverse events, procedures, and other study data should be documented in organizational or study-specific procedures or guidelines [I].11 This is particularly relevant in medical coding because coding tasks are often shared between algorithms and humans in processes that leverage autoencoding technology followed by use of a human coder to handle those terms not codable by the algorithm.
EDC system functionality for medical coding varies considerably with three main process variants. The first (Figure 2a) involves use of type ahead functionality on verbatim term fields to facilitate use of the controlled terminology by the data enterer at the clinical investigational site. Because controlled terminologies can be very large, for example MedDRA or SNOMED contain between 70,000–100,000 terms, this model requires optimized architecture and infrastructure to assure adequate system response time. Further, the structure of controlled terminologies can vary widely from a list to a single hierarchy taxonomy to a poly-hierarchical taxonomy to a poly-hierarchical system of multiple relationships, i.e., an ontology. Supporting this model means that the EDC system also must have functionality to parse these controlled terminologies for terms, to store them, and to update them with new releases of the terminology. For these reasons, while the model in Figure 2a, i.e., having sites review automatically applied codes may be attractive, it is not often the case. A second model involves the type ahead and dictionary management functionality described in 2a, but also allows for non-matching verbatim terms to be saved and coded later by a central medical coder (Figure 2b). This model requires that the infrastructure for medical coding is available at the time of data entry in addition to functionality to support central coding and issuing coding-related queries to sites through the EDC system. While this model decreases coding-related queries via the type ahead matching and real-time review of the matched term by the sites, it requires the infrastructure of both front-end and back-end coding. The third model is the traditional post-processing model and involves the clinical investigational site entering verbatim terms that are later coded centrally (Figure 2c). This model relieves the pressure of system response time associated with type ahead coding. However, a mechanism of communicating coding-related queries to the sites, preferably through the EDC system interface, is required.
a: Type ahead coding running real-time on the EDC system interface with no processing outside of the EDC system requiring coding-related communication with the clinical investigational sites. b: Type ahead coding running real-time on the EDC system interface with post processing functionality for terms that do not code or other coding-related queries communicated to sites via the EDC system interface. c: All coding done as post processing with coding-related queries communicated to sites via the EDC system interface.
Optimizing the coding quality and system usability by the clinical investigational sites requires a good understanding of the capability of the EDC system to support one or more of the coding models.
If the EDC system is capable of handling coding, the sponsor should decide whether the user should be able to see coded terms or only the reported verbatim terms. [VI] Unless coded terms are included in queries, to avoid confusion, it is recommended not to display coded terms back to the site user. [VI] Clinical data management should work with team members trained in controlled clinical terminology to determine how data coding should be handled. [VI] Ensure the clinical team understands who will be coding terms that do not match or otherwise auto-encode and how clinical review of coding, where deemed necessary, will occur. [VI] Documentation of the coding process should include training or guidelines for assigning codes, the frequency of coding and clinical review, procedures, timing for any data imports or exports required, and management of dictionaries used in the coding process.
Most terms that code will have additional codes/values associated, as such a mechanism to re-associate the codes with this additional information should be built into the database. For example, a MedDRA term will always have an associated System Organ Class (SOC).
19) Developing and Testing a Study within an EDC System
EDC functionality with respect to building a study varies widely. Some systems require computer programs to be written to create data entry screens and the corresponding logical structures in which data are stored. However, most EDC systems have tools that decrease or altogether eliminate custom programming to set up entry screens and data storage. For example, some EDC systems accept a spreadsheet of data elements by screen and their properties such as the data type, whether a response is required, the prompt to be displayed on the screen, the data collection structure to be used, structure-specific specification of valid values, preceding data element on the screen, and grouping to which the data element belongs. The EDC system then builds the screen according to the spreadsheet. Other systems offer less automation and sometimes more flexibility in screen set-up through using graphical user interfaces where different data collection structures are added to a screen and properties are added to the data collection structure. Similarly, but often to a lesser extent, most EDC systems have tools to facilitate importing and exporting data as well as for the development of edit checks and other rule-based system features such as dynamic visits, forms and fields, screen tab order, and skip patterns.
To the extent custom computer programming is required, professionals trained in relevant programming language, style, and tools are required [I].12 Further, to the extent that custom programming is required, so is a documented process for specification, development, and validation of programmed components [I].12
User testing with comprehensive test cases is strongly recommended for EDC studies. [VI] Because the users are external, problems can be more impactful and harder to remediate than problems in a system used by internal data management staff. Errors in rule specification can cause equally serious problems such as rules never firing or firing in false positive manner. Fixing problems with rules often requires site users to go back and address newly fired discrepancies on data previously thought complete and clean. For this reason, each rule should be tested with at least one boundary with a case that causes the rule to fire and a test case that should not cause the rule to fire. [VI] Where rules are tested in a manner that does not address each logic path in the rule, rules should be monitored once in production to identify rules that fire too frequently and rules that have not yet fired. [VI] The more data are accrued, the better the ability of such monitoring to identify rules likely to be malfunctioning. Active monitoring finds problems sooner and prevents sites from receiving queries from errant rules fixed late in the study. Studies should not collect production data until User Acceptance Testing (UAT) has been performed and documented. [VI] The extent of UAT, i.e., the number and type of test cases for screens and rules, can and should be risk-based. [VI]
While tools and functionality obviating custom programming can save time and resources, they do not eliminate the need for testing. A system with absolutely no custom programming in study set-up should be tested. [VI] This is because errors can occur during set up and unintended consequences can result from errors in set-up. For example, a spreadsheet listing fields to be displayed on a screen can contain an error in the data type, prompt, data collection structure, or valid values. Such errors result in systematic data quality problems because they most often impact every value entered in the affected field. Further, a system that functions perfectly according to specifications can cause unintended problems once in use by humans and at multiple institutions. For example, the set-up specifications for a study on which data were to be entered in-house contained different response order for questionnaire data and for clinical observations. The inconsistent display order of yes/no radio buttons in a study resulted in an error rate of over 200 errors per 10,000 fields.45 The problem was discovered when the study chair and statistician reviewed the draft tables, found a particular result clinically unlikely, and investigated.45 An astute tester may have detected the problem before the system was released. Other unintended problems include a screen so long it requires scrolling causing sites to miss fields at the bottom, another field layout that causes fields to be consistently missed, and misleading prompts that cause inconsistent data entry. Testing in-house may catch some problems. Testing at investigational sites will likely catch more problems. Thus, some testing of “zero-programming” or configuration-only set-up is recommended. [VI]
Regardless of the type and amount of testing done, observing a system’s operation once in production is recommended. [VI] System observation can take many forms including review of system error logs, distributional and conditional comparison of entered data, queries, query response, and operational metadata across visits, forms, data elements, sites, and users. Routine and ongoing system observation may serve as a trigger for risk-based activities including site calls, monitoring, investigation, and auditing. These activities also help meet the intent of ICH E6 section 5.1.3, “Quality control should be applied to each stage of data handling to ensure that all data are reliable and have been processed correctly”.11 Most importantly, frequent ongoing observation is proactive and catches problems earlier than relying on downstream processes to identify things that look odd, for example, during analysis programing or table and listing review. “Ironically, there is a major difference between a process that is presumed through inaction to be error-free and one that monitors mistakes. The so-called error-free process will often fail to note mistakes when they occur.”46 For these reasons, ongoing systematic observation of system performance is recommended. [VI] Such monitoring may itself be risk-based in terms of the frequency and extent of the observations and the type of items monitored. [VI]
20) Study Start
End User Preparation (Site)
Good clinical practices advise site assessments. In addition to reinforcing the Title 21 CFR Part 11 requirement that individuals involved in conducting a trial should be qualified by education, training, and experience to perform their respective task(s), the introduction to quality management section 5.0 of E6(R2)states that the, “sponsor should implement a system to manage quality throughout all stages of the trial process” and is followed by a description of risk identification and control.11,12 In that same section 5.0.2, risk identification, states that the, “sponsor should identify risks to critical trial processes and data”, that risks should be considered at both the system and trial levels.11 To meet the intent of regulation and guidance, a site assessment should confirm a site’s ability to access and use the EDC system prior to initiation of the study at the site. [VI] Such an assessment may include personnel qualification prior training and experience, institutional infrastructure, and system training and demonstration of competence in preparation for a study.
As part of operating a validated system, the sponsor or designee is responsible for ensuring that sites are qualified to use hardware or software required by the EDC system.12 In many parts of the world, access to the internet and associated infrastructure are almost wholly ubiquitous; however, there may still be sites that have connectivity, hardware, or software challenges. For example, a site’s internet browser or browser version, may not be compatible with the EDC system, or the local area may have less than ideal electrical power quality. Internet-based test sites will suffice in many situations; i.e., “if you can access this site, you will be able to use the EDC system”. In rural areas or parts of the world lacking consistent electrical power or internet access, more consideration should be given to a site’s ability to use EDC. Site evaluation and qualification with respect to EDC systems by the sponsor or designee should occur during start-up activities prior to subject screening. [VI]
21) EDC Account Management
Setting System Rights Determined by Roles and Privacy
Title 21 CFR Part 11 requires, “Limiting system access to authorized individuals” including use of, “authority checks to ensure that only authorized individuals can use the system, electronically sign a record, access the operation or computer system input or output device, alter a record, or perform the operation at hand”12. System access and privileges within the system need to be considered for all roles using the EDC system. Management of system access and privileges begins with enumeration of the roles and the responsibilities and tasks to be associated for each role within the EDC system. [VI] Available roles, tasks, and allowed associations vary across EDC systems. Factors to be considered when defining user roles include the following:
Data Entry Rights—It is important to understand which users will need access to each form or groups of forms within the study. In most clinical trials, site users will be the most common user who will need data entry permissions; however, in some studies, call center, central reading center, and core lab users or patients entering self-reported data may need more limited data entry rights. In some scenarios the sponsor or their designee’s staff may need entry or edit rights. For example, in EDC systems with limited coding functionality, dictionary coding requires that sponsor staff be able to enter or modify verbatim term fields on a form. To ensure that integrity and reliability of data are maintained, sponsors should carefully consider which fields will be modifiable by the sponsor team.
Data Management Review (DM Review) or other custom rights—Some EDC systems are configured to have other workflows such as DM Review, Medical Monitor (MM) review, etc. If these workflows are available as part of the EDC system and turned on for a particular study, it is imperative to have certain users set up with the appropriate permissions and process documentation outlining the workflow and necessary steps.
Source Data Verification (SDV) rights—Clinical Research Associates (CRAs) or other clinical operations staff may have SDV rights to indicate source-verified fields and to enter queries to the site where discrepancies are noted.
Read-only access—Some roles may require read access to some fields; for example, a research pharmacy filling an order or a central reading center viewing data associated with an event under review.
Creating manual queries—CDM, CRAs, Drug Safety, medical coders, etc. may all have the ability to create different types (CRA, DM, etc.) of manual queries.
Answering or resolving queries (manual or system)—Sites will always have the ability to answer manual or system queries, but some EDC systems may allow other configured users (DM, Drug Safety, etc.) to respond to queries as a part of the data cleaning process.
Closing queries (manual or system) — The roles with the rights to close queries is an organizational decision. CRAS may only be able to close or resolve queries created by a CRA user group, while CDMs can close system or manual DM queries after reviewing site responses. In some EDC configurations, CRAs and DMs could share responsibility for closing one another’s queries.
Report creation, generation, or view-only access at both the site and by the sponsor or designee should be considered. Some possible scenarios include limiting access so that each site can only generate reports for their subjects or CRAs can generate reports for subjects at their sites or the entire study depending on the user permissions, limiting report generation across countries or regions, or limiting report creation to CDM staff who have received more advanced training.
Data extraction should be similarly limited to prevent unintended disclosure of data.
Some EDC systems offer permissions to database creation.
Documentation and tracking over time of access and privileges in the system supports auditability of procedures.
22) User IDs and Passwords
User credentials such as user identifiers and passwords are essential to the control required for non-repudiation by Title 21 CFR Part 11. As such, Part 11 section 11.10 requires the “establishment of, and adherence to, written policies that hold individuals accountable and responsible for actions initiated under their electronic signatures, in order to deter record and signature falsification”. Part 11 section 11.100 requires written certification to the FDA that electronic signatures, “are intended to be the legally binding equivalent of traditional handwritten signatures” [I].12 In addition, electronic signatures must be unique to one individual and should not be reassigned and the identity of individuals using electronic signatures must be verified [I].12
Processes for dissemination of user credentials such as user identifiers and passwords should be established. [VI] These processes should include tracking that users have been properly trained prior to receiving access to the system [I].12 To support non-repudiation by keeping user credentials secure, the EDC system should force users to change their password at first log-in. [VI] Training or system documentation should educate users as to the rules and regulations regarding keeping user ID and password information confidential, as well as requirements for changing their passwords. [VI] Lastly, the training materials should instruct users on what to do should they lose or forget their ID and/or password. [VI] Thus, site users should have an individual and not a shared email account to receive user IDs and passwords for EDC applications. [VI] Some institutional sites may use a shared email account for operational purposes. This can be problematic if the EDC system uses email address to uniquely identify user accounts.
23) Account Management
The account management process may be defined with cross-functional input and should be maintained by a function with knowledge of and close communication with the sites. [VI] This supports site user training as well as validation of an individual’s identity and detecting personnel changes requiring changes in system access and privileges. Consideration could be given to linking the CTMS to the account creation and activation system, thereby eliminating the need to transfer user information between systems. [VI] A secure process for managing access and privileges will minimize the number of manual steps that are included and employ separation of duties. [VI] An example of a typical account activation process is enumerated below.
A user is trained and authorized to be granted access to the system for a specific role.
The sponsor or designee confirms that EDC training has been completed by the user.
An account is created, and access provided.
Account use is monitored for aberrant behavior and site staffing is monitored for changes necessitating discontinuation of access and onboarding new site personnel.
Accounts are disabled as the access need diminishes when individual patients, visits, or the database are locked.
24) Training Prior to System Access
Title 21 CFR Part 11 requires a determination that, “persons who develop, maintain, or use electronic record/electronic signature systems have the education, training, and experience to perform their assigned tasks” [I].12 Following a risk-based approach, training for site users with previously established system education, training, or experience may be less extensive than for site users lacking relevant education, previous training, or experience with the system. Similarly, training for an open-label extension or similar trial with similar data collection in the same system may be significantly reduced. On the other hand, for inexperienced sites or new system functionality or processes, study-specific training in the EDC system may be more extensive and include an assessment of competence. [VI]
Documentation of training completion or its location should be maintained in the Trial Master File (TMF), even though it may also be maintained in the EDC training system [I].11 Training documentation may also be given to trainees and used to support qualification on future studies. [VI]
User training on both the system and study application is important. There are varying views on the extent to which these two components should be included in training. At a minimum, all users should have competency in basic system functionality available through their permissions. For a site user, these usually include how to login, how to navigate to patients and visits in the system, how to enter and update data, and how to respond to system generated data discrepancy notices and manual data discrepancy. [VI] Often, studies add dynamic behavior; in these cases, study specific training covering how the study eCRF responds to different user actions and input may be required. Because of the increased interaction between data and form behavior in EDC it may be effective to combine training on the study eCRF with training on data collection such as training on guidelines for where in the medical record to find needed data and what value should be chosen in the case of multiple conformant values. [VI]
User training can be provided through different methods, including
Self-study of reading or e-learning materials followed by demonstration of competency using sample forms in a training environment
Demonstrating competency in training environments that provide training exercises with examples that are generic or customized to the study-specific workflow
Web-based instruction or decentralized/remote demonstration followed by demonstration of competency using sample forms in a training environment
Face-to-face training for users in a central training facility, such as at investigators’ meetings or other centralized training meetings.
Consideration should be given to issues posed by language barriers to training. For example, investigator meetings could provide simultaneous translation for all languages spoken by participants, a train the trainer strategy could be employed, or training materials could be translated into the users’ native languages.
The training requirements articulated in Part 11 also apply to individuals who build, test, and maintain the study eCRF and those who manage accounts, privileges, and study data within the EDC system [I].12 Individuals with these responsibilities should have documented training corresponding to their roles and responsibilities [I].12
25) Study Considerations and Start-up Timelines
One-third of companies responding to the eClinical Landscape survey reported “often” or “always” releasing the study-specific database after the First Patient First Visit (FPFV).9 In the survey, release the EDC system after enrollment had begun was associated with significantly longer data entry time and a longer time from Last Patient Last Visit (LPLV) to database lock.9 Further, “always” releasing the EDC after FPFV was associated with data management cycle time metrics nearly double those for companies reporting “never” doing so.9 Starting enrollment of a study prior to a complete EDC study-build is strongly discouraged [III].8,9,19,20,21,23 Starting a study prior to the complete EDC study-build diminishes the advantage of using EDC. Much of EDC Chapter 3, “Electronic Data Capture – Study Conduct, Maintenance, and Closeout, focuses on leveraging the EDC system to manage a study and provides additional support and rationale for this recommendation”.
To minimize time required for system development, the set-up of the EDC system should be managed as a project in and of itself and as a key study milestone. [VI]
a) Sponsor/CRO EDC Vendor Responsibilities
Though implementation of the study application may be performed by contracted vendors, the sponsor is ultimately responsible for the adherence to regulatory considerations, and final acceptance of the study implementation. A sponsor may choose to build the study in-house, using tools provided by an EDC Vendor, or outsource the build to a third party such as a CRO or independent contractor. In some cases, the EDC vendor may be contracted for the study build. When several companies are involved with the database build, it is still necessary for them to have frequent communication and guidance from the sponsor. At a minimum, the sponsor should retain signatory approval of the EDC build or components of the EDC build such as the eCRF design, edit checks and testing. As part of initiation on production use of the system, documentation of the aforementioned activities should be stored in the Trial Master File. [VI]
b) International Study Considerations
EDC systems are routinely used in international studies. Many EDC systems have the ability for presenting the EDC interface in multiple languages or collecting the data in multiple languages. CDMs should work with stakeholders to understand language and time zone needs of the study or any components of the eCRF. Issues to consider include the following:
Whether the local language can be used in a multi-national study. Many coordinators speak more than one language. Asking this simple question or challenging the status quo in this area can avoid unnecessary work.
Planning enough time for eCRFs that will be translated, rendered in multiple languages, and undergo back-translation.
Ensuring that the eCRF completion guidelines are available in appropriate languages.
Understanding how time zone differences will affect time and date stamping of the EDC audit trail, and external data that may be collected in other time zones.
Consideration of the wording of electronic and manual queries to ensure they will be understood by speakers of other languages.
Ensuring that helpdesk support has sufficient language coverage to assist sites with system issues in their local language and time zones.
Understanding how data collected in different languages will be interpreted and used for analysis.
26) Recommended Standard Operating Procedures
a) Sponsor (or designee such as a CRO or EDC Vendor) SOPs
Section 5.0.1 of ICH E6(R2) states that “During protocol development the Sponsor should identify processes and data that are critical to ensure human subject protection and the reliability of trial results.”11 This implies that organizations should map out the processes involved in study design, start-up, conduct, and closeout and make explicit decisions about which are considered to impact human subject protection and the reliability of study results. Organizational processes may be partitioned differently leading to different scope and titles for SOPs. We provide the following as a list of processes commonly considered to impact human subject protection and the reliability of study results. Organizations may differ as to how these processes are covered in SOPs.
Data Management Plan Creation and Maintenance
Document Control (ICH E6 R2 8.0)
Software Development Lifecycle (Title 21 CFR Part 11)
System validation and functionality testing including how study eCRFs will be specified, developed, or configured and tested (Title 21 CFR Part 11, ICH E6 R2 5.5.3 b)
Data collection (ICH E6 R2 5.0)
Data processing including how medical coding, data review and validation, and integration of external data will be handled (ICH E6 R2 5.0)
System maintenance (ICH E6 R2 5.5.3 b)
System change control (ICH E6 R2 5.5.3 b)
System security measures (ICH E6 R2 5.5.3 b)
Data backup and recovery (ICH E6 R2 5.5.3 b)
Contingency planning (ICH E6 R2 5.5.3 b)
System decommissioning (ICH E6 R2 5.5.3 b)
Vendor selection and management (Title 21 CFR 312.52,47 ICH E6 R2 5.0)
User Access Creation, Modification, and Revocation (Title 21 CFR Part 11)
User training and support (Title 21 CFR 312.52, ICH E6 R2 5.0)
Specification, development, and testing of study status reports (ICH E6 R2 5.0)
b) SOPs at Clinical Investigational sites
Title 21 CFR Part 11 section 11.10 (j) states that “The establishment of, and adherence to, written policies that hold individuals accountable and responsible for actions initiated under their electronic signatures, in order to deter record and signature falsification.”12 (Part 11 section 11.10 (j)) In section 11.30 Part 11 states that “Persons who use open systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, as appropriate, the confidentiality of electronic records from the point of their creation to the point of their receipt.”12 and in section 11.300 that “Persons who use electronic signatures based upon use of identification codes in combination with passwords shall employ controls to ensure their security and integrity.”12 and calls out “loss management procedures” for lost or stolen system access credentials. Thus, it has been recommended that sites maintain one or more SOPs describing the following common site processes for using a Sponsor’s EDC system for a study.48
Statement that the site has certified to FDA that, “the electronic signatures in their system, used on or after August 20, 1997, are intended to be the legally binding equivalent of traditional handwritten signatures.” and a process for assuring that site EDC system users are informed that electronic signatures are legally binding. (21 CFR Part 11)
Assuring that each user has a unique user ID and system access credentials (21 CFR Part 11)
Prompt reporting lost or otherwise compromised passwords (21 CFR Part 11)
Use of Sponsor-provided training on EDC systems (21 CFR Part 11)
Use of Sponsor-provided procedures for study EDC system use including data collection, entry, resolution of discrepant data, and EDC system automated decision support or workflow (21 CFR Part 11)
Sponsor notification of new site employees needing EDC training and access (21 CFR Part 11)
Reporting problems with Sponsor-provided EDC systems (21 CFR Part 11)
Assuring prior IRB approval or IRB determination that approval is not needed prior to site use of changes to data to be collected in an EDC system49 (21 CFR Part 56 section 56.109)
Receipt and retention of data entered into Sponsor EDC systems (21 CFR Part 312 section 312.57)
27) Literature Review
This revision is based on a systematic review of the peer-reviewed literature. The goals of this literature review were to (1) identify published research results and reports of EDC methods and evaluation and (2) identify, evaluate, and summarize evidence capable of informing the practice of implementation and start-up of studies using web-based EDC. The following PubMed query was used:
(“electronic data capture” OR “EDC” OR (internet AND “data collection”)) AND (“clinical trial” OR “clinical trials” OR “clinical study” OR registry OR registries OR “observational study” OR “interventional study” OR “phase 1” OR “phase 2” OR “phase 3” OR “phase 4” OR “phase I” OR “phase II” OR “phase III” OR “phase IV” OR “first in man” OR “clinical research” OR “device study” OR “interventional trial” OR “phase 1” OR “phase 2” OR “phase 3” OR “phase 4” OR “phase I” OR “phase II” OR “phase III” OR “phase IV” OR RCT OR “randomized clinical trial” OR “non-interventional” OR “post-marketing authorization” OR “post authorization” OR “adaptive trials” OR “feasibility study” OR “phase 2/3” OR “phase II/III” OR “phase 2a” OR “phase 2b” OR “phase IIa” OR “phase IIb” OR “phase IIb/IIIa” OR “phase 2b/3a”)
The search query was customized for and executed on the following databases: PubMed (777 results); CINAHL (230 results); EMBASE (257 results); Science Citation Index/Web of Science (393 results); Association for Computing Machinery (ACM) Guide to the Computing Literature (115 results). A total of 1772 works were identified through the searches. The latest search was conducted on February 8, 2017. Search results were consolidated to obtain a list of 1368 distinct articles. Because this was the first review for this chapter, the searches were not restricted to any time range.
Two reviewers used inclusion criteria to screen all abstracts. Disagreements were adjudicated by the writing group. Forty-nine sources (mostly articles) meeting inclusion criteria were selected for review. The selected sources were read by the writing group and 109 additional sources identified through the review. Each of these 158 (49 + 109) sources was read for mention of explicit practice recommendations or research results informing practice. A total of 85 sources were deemed relevant to EDC and 73 were excluded by the full text review as not relevant to EDC. Of the 85 relevant sources, 53 were identified as informative for practice in one or more of the EDC GCDMP chapters and 32 were relevant but not informative of practice in any of the three EDC chapters. Twenty-two articles provided evidence for this EDC chapter. Relevant findings from these twenty-two articles have been included in the chapter (Figure 3). This synthesis of the literature relevant to web-based EDC was performed to support the transition of the EDC chapters to an evidence-based guideline.
Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) For Web-based EDC Study Implementation and Start-up.
28) Revision History
| Date | Revision description |
|---|---|
| September 2003 | Initial publication as Electronic Data Capture Principles. |
| May 2007 | Revised for style, grammar, and clarity. Substance of chapter content unchanged. |
| September 2008 | Revised to reflect the orientation of chapter towards the conduct phase of EDC. Content updated and organization of material revised. Study concept and start up, and study closeout content moved to separate chapters. |
| January 2021 | Content updated and organization of material significantly revised. Study implementation and start-up was organized into one comprehensive chapter. |
Competing Interests
The authors have no competing interests to declare.
References
1. Spilker B, Schoenfelder J. Data Collection Forms in Clinical Trials. New York: Raven Press; 1991: Chapters 1–3.
2. Kennedy D, Hutchinson D. CRF Designer. Canary Publications; 2002: ISBN 0-9531174-7-2
3. McFadden E. Management of Data in Clinical Trials. 2nd ed. Hoboken, NJ: John Wiley & Sons; 2007.
4. Avey M. Case report form design. In Rondel RK, Varley SA, Webb CF (eds.), Clinical Data Management. 2nd ed. West Sussex: John Wiley & Sons. 2000; 47–74. DOI: http://doi.org/10.1002/0470846364.ch3
5. Bellary S, Krishnankutty B, Latha MS. Basics of case report form designing in clinical research. Perspect Clin Res. 2014; 5(4): 159–166. DOI: http://doi.org/10.4103/2229-3485.140555
6. Helms RW. Data quality issues in electronic data capture. Drug Inf J. 2001; 35: 827–837. DOI: http://doi.org/10.1177/009286150103500320
7. Stead WW, Lin HS. National Research Council. Computational Technology for Effective Health Care: Immediate Steps and Strategic Directions. Washington, DC: The National Academies Press. 2009. DOI: http://doi.org/10.17226/12572
8. Kush RD, Bleicher P, Kubick WR, et al. eClinical Trials: Planning and Implementation. 1st ed. Boston: Thompson/CenterWatch, Inc.; 2003. ISBN 193062428X, 9781930624283.
9. Wilkinson M, Young R, Harper B, Machion B, Getz K. Baseline assessment of the evolving 2017 eClinical landscape. Ther Innov Regul Sci. 2019; 53(1): 71–80. DOI: http://doi.org/10.1177/2168479018769292. Available at https://pubmed.ncbi.nlm.nih.gov/29714600/.
10. Getz K. Examining Causes of and Potential Solutions to Clinical Data Management Cycle Time Challenges. 2018; 1–10. Boston: Tufts Center for the Study of Drug Development.
11. Food and Drug Administration, US Department of Health and Human Services. ICH E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1), March 2018. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r2-good-clinical-practice-integrated-addendum-ich-e6r1.
12. Food and Drug Administration. US Department of Health and Human Services. Electronic Records; Electronic Signatures, 21 CFR §11 (1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
13. Food and Drug Administration. US Department of Health and Human Services. Guidance for Industry: Computerized Systems Used in Clinical Investigations. May, 2007. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computerized-systems-used-clinical-investigations.
14. Medicines and Healthcare products Regulatory Agency (MHRA) ‘GXP’ Data Integrity Guidance and Definitions, Revision 1. March 2018. Available at https://www.gov.uk/government/publications/guidance-on-gxp-data-integrity.
15. EudraLex: Good Manufacturing Practice (GMP) guidelines, Volume 4: Annex 11: Computerised Systems ( 2011). Available at https://ec.europa.eu/health/documents/eudralex/vol-4_ga.
16. GAMP® 5 A Risk-based Approach to Compliant GxP Computerized Systems. North Bethesda, MD: International Society for Pharmaceutical Engineering (ISPE). 2008.
17. Food and Drug Administration, US Department of Health and Human Services. Guidance for industry: Use of Electronic Health Record Data in Clinical Investigations. July 2018. Accessed September 17, 2020. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-electronic-health-record-data-clinical-investigations-guidance-industry.
18. Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry: Electronic Source Data in Clinical Investigations. September 2013. Accessed August 8, 2018. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electronic-source-data-clinical-investigations.
19. Richardson A. Planning and running the eClinical Trial. Appl Clin Trials. 2003; January.
20. Mitchel JT, Kim YJ, Choi J, et al. The impact of electronic data capture on clinical data management perspectives from the present into the future. MONITOR. 2008; August.
21. Mitchel JT, Kim YJ, Choi J, Hays V, Langendorf J, Cappi S. Impact of IBCTs on clinical trial efficiency. Appl Clin Trials. 2006; August.
22. Mitchel J, Weingard K, Schloss Markowitz J, et al. How direct data entry at the time of the patient visit is transforming clinical research – perspective from the clinical trial research site, INSITE. 2013; 2nd Quarter: 40–43.
23. Litchfield J, Freeman J, Schou H, Elsley M, Fuller R, Chubb B. Is the future for clinical trials internet-based? A cluster randomized clinical trial. Clin Trials. 2005; 2: 72–79. DOI: http://doi.org/10.1191/1740774505cn069oa
24. Summa W. Electronic data capture: automated management of clinical trial data. Pharmind: Special Issue 5a. 2004.
25. Kush R. Electronic data capture-pros and cons. BioExecutive International. 2006; 2(6): S48–52.
26. Laky D. The evolution of EDC into eClinical. epc magazine. 2007; 11(2). Available at: http://www.samedanltd.com/magazine/11/issue/2. accessed September 29, 2020
27. Lu Z. Electronic data-capturing technology for clinical trials, experience with a global postmarketing study. IEEE Eng Med Biol Mag. 2010; 29(2): 95–102. DOI: http://doi.org/10.1109/MEMB.2009.935726
28. Nahm ML, Pieper CF, Cunningham MM. Quantifying data quality for clinical trials using electronic data capture. PLoS ONE. 2008; 3(8): e3049. PMCID: PMC2516178. DOI: http://doi.org/10.1371/journal.pone.0003049
29. Zozus MN, Pieper C, Johnson CM, et al. Factors affecting accuracy of data abstracted from medical records. PLoS ONE. 2015; 10(10): e0138649. PMC4615628. DOI: http://doi.org/10.1371/journal.pone.0138649
30. Zozus MN, Young LW, Simon AE, et al. Training as an intervention to decrease medical record abstraction errors multicenter studies. Stud Health Technol Inform. 2019; 257: 526–539.
31. Zozus NM, Kahn M, Weiskopf N. Clinical research data quality. In Richesson RL, Andrews JE (eds.), Clinical Research Informatics, Health Informatics. 2nd ed. London: Springer-Verlag, 2018. 175–201.
32. Chhabra L, Goel N, Prajapat L, Spodick DH, Goyal S. Mouse heart rate in a human: diagnostic mystery of an extreme tachyarrhythmia. Indian Pacing Electrophysiol J. 2012; 12(1): 32–35. DOI: http://doi.org/10.1016/S0972-6292(16)30463-6
33. Cramon P, Rasmussen AK, Bonnema SJ, et al. Development and implementation of PROgmatic: A clinical trial management system for pragmatic multi-centre trials, optimised for electronic data capture and patient-reported outcomes. Clin Trials. 2014; 11: 344–354. DOI: http://doi.org/10.1177/1740774513517778
34. Zhang J. The nature of external representations in problem solving. Cogn Sci. 1997; 21(2): 179–217. DOI: http://doi.org/10.1016/S0364-0213(99)80022-6
35. Wickens CD, Hollands JG, Banbury S, Parasuraman R. Engineering Psychology & Human Performance. 4th ed. New York: Routledge; 2016.
36. Earley SS. Data Management Association (DAMA) – Dictionary of Data Management Framework. New Jersey. Technics Publications, LLC; 2011.
37. Zozus MN. The Data Book: Collection and Management of Research Data. Boca Raton: Taylor & Francis/CRC Press; 2017. DOI: http://doi.org/10.1201/9781315151694
38. Abouelenein S, Williams TB, Baldner J, Zozus MN. Analysis of professional competencies for the clinical research data management profession. Data Basics. 2020; 26(2): 6–21. Available at www.scdm.org.
39. Howells K. e-Clinical integration strategies. Drug Discov Today Technol. 2006; 3(2). DOI: http://doi.org/10.1016/j.ddtec.2006.06.009
40. Handlesman D. Electronic Data Capture: When Will It Replace Paper? SAS News/Features. 2009. Available at https://web.archive.org/web/20091217165516/http:/www.sas.com/news/feature/hls/sep05edc.html.
41. Haak D, Page CE, Reinartz S, Kruger T, Deserno TM. DICOM for clinical research: PACS-integrated electronic data capture in multi-center trials. J Digit Imaging. 2015; 28: 558–566. Available at https://link.springer.com/article/10.1007/s10278-015-9802-8. DOI: http://doi.org/10.1007/s10278-015-9802-8
42. Franklin JD, Guidry A, Brinkley JF. A partnership approach for electronic data capture in small-scale clinical trials. J Biomed Inform. 2011; 44: S103–S108. DOI: http://doi.org/10.1016/j.jbi.2011.05.008
43. Labovitz GH, Chang YS. Quality Costs: the Good, the Bad and the Ugly. In Labovitz GH, Chang YS, Rosansky V. Making Quality Work: A Leadership Guide for the Results-Driven Manager. Hoboken. John Wiley & Sons, Inc; 1993. Appendix E.
44. Zozus M, Kahn M, Weiskopf, N. Data Quality in Clinical Research. In Richesson R, Andrews J (eds.), Clinical Research Informatics. 2019; 213–248. Available at https://www.springer.com/gp/book/9783319987781. DOI: http://doi.org/10.1007/978-3-319-98779-8_11
45. Nahm M. Data gone awry. Data Basics. 2008; 13(3): 3–7. Available at www.scdm.org.
46. Arndt S, Tyrell G, Woolson RF, Flaum M, Andreasen NC. Effects of errors in a multicenter medical study: preventing misinterpreted data. J Psychiatr Res. 1994; 28(5): 447–459. DOI: http://doi.org/10.1016/0022-3956(94)90003-5
47. Food and Drug Administration. US Department of Health and Human Services. Investigational New Drug Application, 21 CFR §312.52 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.52.
48. Rusnak E. SOPs for electronic records and data collection in clinical research. Issues in clinical trials management. Research Practitioner. 2001; 2(5).
49. Food and Drug Administration. US Department of Health and Human Services. Institutional Review Boards, 21 CFR §56 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=56.