
1) Learning Objectives
After reading this chapter, the reader should understand
the regulatory basis for practices in EDC study conduct, maintenance, and closeout
special considerations for ongoing data surveillance and use when using web-based EDC
common processes used in study conduct of EDC-based studies
methods for managing system access and privileges
common practices for EDC system maintenance and change control during studies
special considerations for close-out of EDC-based studies
2) Introduction
Electronic resources for clinical data management have developed over the last 40 years as a suite of processes and tools to enhance the management, quality control, quality assurance, and archiving of clinical trial research data. This development has led to a major paradigm shift in CDM, with form-based data capture available via the internet to investigator sites.1 These tools and systems have changed the way the CDM team approaches data collection, data validation, data transfer, data analysis, reporting, security, archiving, and storage.
Pre-production activities and planning such as those covered in Chapter 2, “Electronic Data Capture – Implementation and Study Start-up” are necessary for a study employing EDC technology. Equally important is how the technology is used every day to support the ongoing conduct, maintenance, and ultimately closeout of a study. Well-executed conduct and closeout activities are crucial, especially for clinical research data used to support research conclusions, evidence changes in clinical practice, or presented for use in regulatory decision-making.
The integrated addendum to Good Clinical Practice (R2) recommends a Quality Management System (QMS) approach to assuring the safety of human subjects and the integrity of study data.2 Both human safety and data integrity require expertise in ongoing data surveillance to detect problems early and use of the data to inform and support corrective action. These capabilities are designed into the EDC study build as described in Chapter 2, “Electronic Data Capture – Implementation and Study Start-up”. To achieve the benefit, processes must be in place to direct human response to alerts, automated workflow, and lists of potential problems requiring action. For example, a written procedure may be in place that stipulates an easily accessible report of protocol violations by site and describes the actions required by study personnel to follow-up to mitigate risk or prevent future similar occurrences at the site or across sites. The procedure sets the expectation and provides direction for closing the loop on the protocol violations. There is little in the literature about such methods and practices for process design in clinical studies; however, they are the mechanism through which EDC technology improves the conduct of studies, increases the quality of study data, and decreases the elapsed time for data collection and management.
3) Scope
This chapter focuses on EDC-assisted methods for active study management such as problem detection, data reviews, trend analyses, use of work lists, and using the EDC system for common and high-volume communication. Maintaining data security and change control are also addressed as are mid-study data requests and interim analyses. EDC study closeout practices such as final source document review, managing database lock, audits, generation of archival copies, site close-out, and hardware disposal complete the treatment of EDC in clinical studies.
Many of the tasks described in this chapter may be joint responsibilities or performed by individuals outside the Clinical Data Management group. The CDM profession is defined ultimately by the tasks necessary to assure that data are capable of supporting research conclusions. As such, while responsibilities vary across organizations, where CDMs do not directly perform these tasks, the role usually has some organizational responsibility for assuring that data-related tasks have been performed with the rigor necessary to maintain data integrity and support reuse of the data during or after the study.
Detailed information describing design, planning, and pre-production activities for EDC-based studies can be found in Chapter 2, “Electronic Data Capture – Implementation and Study Start-up”. This chapter picks up after study start-up and covers aspects specific to web-based EDC of the active enrollment and data collection phase through to database lock and archival. The Data Management Planning chapter covers general aspects of data collection and management applicable to EDC and other data sources. This chapter covers issues pertaining to study conduct, maintenance, and closeout that are specific to EDC.
4) Minimum Standards
As a mode of data collection and management in clinical studies, EDC systems have the potential to impact human subject protection as well as the reliability of trial results. Regulation and guidance are increasingly vocal on the topic.
The ICH E6(R2) Good Clinical Practice: Integrated Addendum contains several passages particularly relevant to use of EDC systems in clinical studies.2
Section 2.8 “Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective tasks.”
Section 2.10, “All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification.”
Section 5.0 states, “The methods used to assure and control the quality of the trial should be proportionate to the risks inherent in the trial and the importance of the information collected.”
Section 5.1.1 states, “The sponsor is responsible for implementing and maintaining quality assurance and quality control systems with written SOPs to ensure that trials are conducted and data are generated, documented (recorded), and reported in compliance with the protocol, GCP, and the applicable regulatory requirement(s).” Section 5.1.3 states, “Quality control should be applied to each stage of data handling to ensure that all data are reliable and have been processed correctly.”
Section 5.5.1, “The sponsor should utilize appropriately qualified individuals to supervise the overall conduct of the trial, to handle the data, to verify the data, to conduct the statistical analyses, and to prepare the trial reports.”
Section 5.5.3 “When using electronic trial data handling and/or remote electronic trial data systems, the sponsor should: a) Ensure and document that the electronic data processing system(s) conforms to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e., validation).”
Section 5.5.3 addendum “The sponsor should base their approach to validation of such systems on a risk assessment that takes into consideration the intended use of the system and the potential of the system to affect human subject protection and reliability of trial results.” and in the addendum b) states the requirement, “Maintains SOPs for using these systems.”
Section 5.5.3 addendum c-h introductory statement states, “The SOPs should cover system setup, installation, and use. The SOPs should describe system validation and functionality testing, data collection and handling, system maintenance, system security measures, change control, data backup, recovery, contingency planning, and decommissioning.”
Section 5.5.4 under Trial Management, Data Handling and Recordkeeping states that “If data are transformed during processing, it should always be possible to compare the original data and observations with the processed data.”
Similar to ICH E6 R2, Title 21 CFR Part 11 also states requirements for traceability, training and qualification of personnel, and validation of computer systems used in clinical trials.3 Requirements in 21 CFR Part 11 Subpart B are stated as controls for closed systems (21 CFR Part 11 Sec. 11.10), controls for open systems (21 CFR Part 11 Sec. 11.30), signature manifestations (21 CFR Part 11 Sec. 11.50), signature/record linking (21 CFR Part 11 Sec. 11.70). Requirements for electronic signatures are provided in 21 CFR Part 11 Subpart C.
Recommendations in Section A of the 2007 Guidance for Industry Computerized Systems Used in Clinical Investigations (CSUCI) state that “Each specific study protocol should identify each step at which a computerized system will be used to create, modify, maintain, archive, retrieve, or transmit source data.”4
Section B of the CSUCI guidance echoes requirements in Title 21 CFR Part 11, “Standard Operating Procedures (SOPs) pertinent to the use of the computerized system should be available on site” by reiterating that “There should be specific procedures and controls in place when using computerized systems to create, modify, maintain, or transmit electronic records, including when collecting source data at clinical trial sites” and that “the SOPs should be made available for use by personnel and for inspection by FDA.” Thus, comprehensive procedures for use of the computerized systems, i.e., including procedures for system setup or installation, data collection and handling, system maintenance, data backup, recovery, and contingency plans, computer security and change control, whether site or sponsor provided, should be available to the sites at all times.
Section C reiterates document retention requirements under 21 CFR 312.62, 511.1(b)(7)(ii) and 812.140.5,6,7 Further, section C of CSUCI goes on to state, “When source data are transmitted from one system to another …, or entered directly into a remote computerized system … or an electrocardiogram at the clinical site is transmitted to the sponsor’s computerized system, a copy of the data should be maintained at another location, typically at the clinical site but possibly at some other designated site.” It further states, “copies should be made contemporaneously with data entry and should be preserved in an appropriate format, such as XML, PDF or paper formats.”4
Section D further specifies 21 CFR Part 11 principles with respect to limiting access to CSUCI, audit trails, and date and time stamps.
Section E likewise provides further detail regarding expectations for security; e.g., “should maintain a cumulative record that indicates, for any point in time, the names of authorized personnel, their titles, and a description of their access privileges” and recommends that “controls be implemented to prevent, detect, and mitigate effects of computer viruses, worms, or other potentially harmful software code on study data and software.”
Section F addresses direct entry of data including automation and data standardization; data attribution and traceability including explanation of “how source data were obtained and managed, and how electronic records were used to capture data”; system documentation that identifies software and hardware used to “create, modify, maintain, archive, retrieve, or transmit clinical data”; system controls including storage, back-up and recovery of data; and change control of computerized systems.
Section G addresses training of personnel as stated in 21 CFR 11.10(i) that those who “develop, maintain, or use computerized systems have the education, training and experience necessary to perform their assigned tasks”, that training be conducted with frequency sufficient to “ensure familiarity with the computerized system and with any changes to the system during the course of the study” and that “education, training, and experience be documented”.
The Medicines & Healthcare products Regulatory Agency (MHRA) ‘GXP’ Data Integrity Guidance and Definitions covers principles of data integrity, establishing data criticality and inherent risk, designing systems and processes to assure data integrity, and also covers the following topics particularly relevant to EDC.8
Similar to ICH E2 (R2), MHRA Section 2.6 states that “Users of this guidance need to understand their data processes (as a lifecycle) to identify data with the greatest GXP impact. From that, the identification of the most effective and efficient risk-based control and review of the data can be determined and implemented.”
Section 6.2, Raw Data states that “Raw data must permit full reconstruction of the activities.”
Section 6.7 Recording and Collection of Data states that “Organisations should have an appropriate level of process understanding and technical knowledge of systems used for data collection and recording, including their capabilities, limitations and vulnerabilities” and that “The selected method [of data collection and recording] should ensure that data of appropriate accuracy, completeness, content and meaning are collected and retained for their intended use.” It further states that “When used, blank forms … should be controlled. … [to] allow detection of unofficial notebooks and any gaps in notebook pages.”
Section 6.9 Data Processing states that “There should be adequate traceability of any user-defined parameters used within data processing activities to the raw data, including attribution to who performed the activity.” and that “Audit trails and retained records should allow reconstruction of all data processing activities…”
The General Principles of Software Validation; Final Guidance for Industry and FDA Staff (2002) provides guidance regarding generally recognized software validation principles which can be applied to any software, inclusive of that used to support clinical trials.9
Section 2.4 “All production and/or quality system software, even if purchased off-the-shelf, should have documented requirements that fully define its intended use, and information against which testing results and other evidence can be compared, to show that the software is validated for its intended use.”
Section 4.7 (Software Validation After a Change) “Whenever software is changed, a validation analysis should be conducted, not just for validation of the individual change, but also to determine the extent and impact of that change on the entire software system”
Section 5.2.2 „Software requirement specifications should identify clearly the potential hazards that can result from a software failure in the system as well as any safety requirements to be implemented in software.”
Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Volume 4, Annex 11): Computerised Systems provides the following guidelines when using computerized systems in clinical trials.10 Though the guidance is in the context of manufacturing, it is included to emphasize the consistency of thinking and guidance relevant to use of computer systems in clinical trials across the regulatory landscape.
Section 1.0 “Risk management should be applied throughout the lifecycle of the computerised system taking into account patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerised system.”
Section 4.2 “Validation documentation should include change control records (if applicable) and reports on any deviations observed during the validation process.”
Section 4.5 “The regulated user should take all reasonable steps, to ensure that the system has been developed in accordance with an appropriate quality management system.”
Section 7.1 “Data should be secured by both physical and electronic means against damage. Stored data should be checked for accessibility, readability, and accuracy. Access to data should be ensured throughout the retention period.”
Section 7.2 “Regular back-ups of all relevant data should be done. Integrity and accuracy of backup data and the ability to restore the data should be checked during validation and monitored periodically.”
Section 9.0 “Consideration should be given, based on a risk assessment, to building into the system the creation of a record of all GMP-relevant changes and deletions (a system generated “audit trail”). For change or deletion of GMP-relevant data the reason should be documented. Audit trails need to be available and convertible to a generally intelligible form and regularly reviewed.”
Section 10.0 “Any changes to a computerised system including system configurations should only be made in a controlled manner in accordance with a defined procedure.”
GAMP 5: A Risk-based Approach to Compliant GxP Computerized Systems (2008) suggests scaling activities related to computerized systems with a focus on patient safety, product quality and data integrity.11 It provides the following guidelines relevant to GxP regulated computerized systems including systems used to collect and process clinical trial data:
Section 2.1.1 states, “Efforts to ensure fitness for intended use should focus on those aspects that are critical to patient safety, product quality, and data integrity. These critical aspects should be identified, specified, and verified.”
Section 4.2 “The rigor of traceability activities and the extent of documentation should be based on risk, complexity, and novelty, for example a non-configured product may require traceability only between requirements and testing.”
Section 4.2 “The documentation or process used to achieve traceability should be documented and approved during the planning stage, and should be an integrated part of the complete life cycle.”
Section 4.3.4.1 “Change management is a critical activity that is fundamental to maintaining the compliant status of systems and processes. All changes that are proposed during the operational phase of a computerized system, whether related to software (including middleware), hardware, infrastructure, or use of the system, should be subject to a formal change control process (see Appendix 07 for guidance on replacements). This process should ensure that proposed changes are appropriately reviewed to assess impact and risk of implementing the change. The process should ensure that changes are suitably evaluated, authorized, documented, tested, and approved before implementation, and subsequently closed.”
Section 4.3.6.1 “Processes and procedures should be established to ensure that backup copies of software, records, and data are made, maintained, and retained for a defined period within safe and secure areas.”
Section 4.3.6.2 “Critical business processes and systems supporting these processes should be identified and the risks to each assessed. Plans should be established and exercised to ensure the timely and effective resumption of these critical business processes and systems.”
Section 5.3.1.1 “The initial risk assessment should include a decision on whether the system is GxP regulated (i.e., a GxP assessment). If so, the specific regulations should be listed, and to which parts of the system they are applicable. For similar systems, and to avoid unnecessary work, it may be appropriate to base the GxP assessment on the results of a previous assessment, provided the regulated company has an appropriate established procedure.”
Section 5.3.1.2 “The initial risk assessment should determine the overall impact that the computerized system may have on patient safety, product quality, and data integrity due to its role within the business processes. This should take into account both the complexity of the process, and the complexity, novelty, and use of the system.”
The FDA guidance, Use of Electronic Health Record Data in Clinical Investigations, emphasizes that data sources should be documented and that source data and documents be retained in compliance with 21 CFR 312.62(c) and 812.140(d).12
Section V.A states, “Sponsors should include in their data management plan a list of EHR systems used by each clinical investigation site in the clinical investigation” and, “Sponsors should document the manufacturer, model number, and version number of the EHR system and whether the EHR system is certified by ONC”.
Section V.I states, “Clinical investigators must retain all paper and electronic source documents (e.g., originals or certified copies) and records as required to be maintained in compliance with 21 CFR 312.62(c) and 812.140(d)”.
Similarly, the FDA’s guidance, Electronic Source Data Used in Clinical Investigations recommends that all data sources at each site be identified.13
Section III.A states that each data element should be associated with an authorized data originator and goes on to state, “A list of all authorized data originators (i.e., persons, systems, devices, and instruments) should be developed and maintained by the sponsor and made available at each clinical site. In the case of electronic, patient-reported outcome measures, the subject (e.g., unique subject identifier) should be listed as the originator.”
Section III.A.3 elaborates on Title 21 CFR Part 11 and states, “The eCRF should include the capability to record who entered or generated the data [i.e., the originator] and when it was entered or generated.” and “Changes to the data must not obscure the original entry, and must record who made the change, when, and why.”
Section III.A.5 states that the FDA encourages “the use of electronic prompts, flags, and data quality checks in the eCRF to minimize errors and omissions during data entry”.
Section III.C states, “The clinical investigator(s) should retain control of the records (i.e., completed and signed eCRF or certified copy of the eCRF).” In other words, eSource data cannot be in sole control of the sponsor.
As such, we state the following minimum standards for study conduct, maintenance, and closeout using EDC systems.
5) Best Practices
Best practices were identified by both the review and the writing group. Best practices do not have a strong requirement based in regulation or recommended approach based in guidance, but do have supporting evidence either from the literature or consensus of the writing group. As such best practices, like all assertions in GCDMP chapters, have a literature citation where available and are always tagged with a roman numeral indicating the strength of evidence supporting the recommendation. Levels of Evidence are outlined in Table 3.
Minimum Standards.
| 1. | Establish and follow SOPs that include EDC specific aspects of study conduct, maintenance, and closeout.2 [I] |
| 2. | Document the source for data and changes in sources of data at each site including explicit statement that the EDC system is used as the source where this is the case.13 [I] |
| 3. | Ensure data values can be traced from the data origination through all changes and that the audit trail is immutable and readily available for review.3 [I] |
| 4. | Establish and follow SOPs for change control (and documentation thereof) for changes to the underlying EDC system and the study-specific EDC application including the eCRF, data processing, and other dynamic system behavior.3 [I] |
| 5. | Assure procedures for use of computerized systems at clinical sites are in place and available to the site personnel at all times, including procedures for system setup or installation, data collection and handling, system maintenance, data backup, recovery, and contingency plans, computer security, and change control.3 [I] |
| 6. | Complete testing prior to implementation and deployment to sites.3 [I] |
| 7. | Establish and follow SOPs to ensure that all users have documented education, experience, or training supporting their qualification for functions relevant to their role prior to using the system; assure that site users receive training on significant changes to the study-specific EDC application.3 [I] |
| 8. | Establish and follow SOPs to limit data access and permissions to authorized individuals and to document data access and permissions.3 [I] |
Best Practices.
| 1. | Data should be entered by the site staff most familiar with the patients and data so that error is reduced and discrepancies identified during entry can be resolved during entry.14,15,16 [VI, VII] |
| 2. | Data flow should be immediate and continuous.17 [V] |
| 3. | Establish and follow procedures to continually surveil and mine study data and metadata using alerts or reports to detect trends and aberrant events, to prioritize and direct correct and for preventative action.17 [V] |
| 4. | Leverage EDC technology to provide decision support, process automation, and connectivity with other information systems used on the study where practical and useful.16 |
| 5. | The EDC system and all intended data operations such as edit checks and dynamic behavior should be in production prior to enrollment of the first patient.17,18,19,20,21 [II, V] |
| 6. | Establish and maintain all-stakeholder study team communication to manage data entry, query resolution, and change control throughout the study. [VI] |
| 7. | The EDC system should be tightly coupled to the safety data collection and handling process with potential events triggered from the EDC system and ongoing synchronicity.17 [V] |
| 8. | Take advantage of opportunities to solicit feedback and provide additional training at investigator meetings, study coordinator teleconferences, and monitoring visits, as well as through communications such as splash screens or other notification functionality available in the EDC. [VII] |
| 9. | Employ active management processes for data acquisition and processing through communication of data entry and query timelines, frequent reporting of status, and regular follow-up to resolve lagging activities so that data are available rapidly to support trial management.18,22,23,24 [V] |
| 10. | Employ ongoing surveillance when new systems or new functions are implemented to assure that the EDC system continues to operate as intended.22 [V] |
| 11. | Manage activities from data origination to “clean” and use an incremental form or casebook lock strategy to lock-as-you-go to reduce the amount of data review and locking needed upon study completion.16 [VII] |
Grading Criteria.
| Evidence Level | Evidence Grading Criteria |
|---|---|
| I | Large controlled experiments, meta, or pooled analysis of controlled experiments, regulation or regulatory guidance |
| II | Small controlled experiments with unclear results |
| III | Reviews or synthesis of the empirical literature |
| IV | Observational studies with a comparison group |
| V | Observational studies including demonstration projects and case studies with no control |
| VI | Consensus of the writing group including GCDMP Executive Committee and public comment process |
| VII | Opinion papers |
6) Working towards Closeout – Data Reviews, Trend Analysis, and Remediation
After all of the work to design, develop, test, and implement an EDC study application, the move to production, also called go-live, can be quite a relief. However, the active enrollment and data collection phase of a study is no time to rest. In fact, when recruitment starts, data management tasks not only change, but in a large and well-managed study they often accelerate. In studies where high data volume is accompanied by high enrollment, managing and controlling the collection and processing of data can consume multiple dedicated people. Without sufficient resources, backlogs of data entry, other data processing, or Source Data Verification (SDV) develop quickly. Backlogs delay access to and use of the data not only for interim analyses but also for monitoring and managing the study, ultimately eroding the value of EDC.21 Thus, working toward database lock should start before the first piece of data is collected.16,23,24 As such, an important component of the Data Management quality management system is the knowledge, tools, and ability to accurately calculate the number of people with the required skills needed to stay abreast of incoming data for a study.25 [VI, VII] Further, processes2 and roles of those collecting, processing, and reviewing data should be well-defined prior to study start.21
Provided good operational design, the next major step is to follow through on that design by conducting the study as planned.21 Conducting a study as planned often benefits from the support of automation, workflow controls, and other tools as described in the previous chapter to alleviate manual tasks where possible and support them in the many cases where full automation is not possible. Examples include immediate decision support, prompts, constraints, reminders, and alerts at the time of data entry.14,26,27,28 Such controls are the mechanism through which EDC systems support and often enforce process standardization across sites and individual users.16 For information about setting up a study within an EDC system, see the GCDMP Chapter 2, “Electronic Data Capture (EDC) – Implementation and Study Start-up”.
The vast majority of benefits of web-based EDC accrue during the active data collection phase of the study. Web-based information systems make centralization of information from multiple locations possible and in real time and, likewise, support decentralized, simultaneous access use of that same information. With web-based EDC systems, immediate oversight and coordination became possible for the first time in clinical trials.15,21,28,29,30,31
The rapid availability of study data in an EDC system allows project teams to detect problems such as delays, discrepancies, and deviations and make decisions earlier than in paper-based studies.23,24 For example, early notice of protocol noncompliance is crucial to study conduct, especially with complex protocols. Algorithms to detect and report systematically identified risk areas should be programmed at the beginning of a study and be run frequently to monitor incoming data for operational problems.16 Analysis of identified problems and mistakes may indicate the need to provide additional training or job aids or to amend the protocol. In addition, ongoing safety data review helps identify trends and alert investigators immediately of patient safety issues during the study, potentially averting harm or saving lives. Although serious adverse event (SAE) notifications can occur rapidly in paper-based studies by phone or fax, an EDC study offers a more automated and systematic approach through alerts, connectivity, and workflow controls.
Interim efficacy and safety data reviews can be performed earlier in an EDC-based study using the most current, near real-time patient information. Non-serious adverse events and other pertinent patient information can be also be reviewed earlier in the study, ensuring that a Data and Safety Monitoring Board (DSMB) has a current, complete picture of the patient safety profile. Decisions by a DSMB to stop a study because of safety concerns or lack of efficacy can be made much more quickly, ensuring better subject protection and lower costs.
Given the advantages of having data immediately after a study participant is enrolled, it is surprising that about one-third of companies responding to the eClinical Landscape survey reported “often” or “always” releasing the study-specific database after the First Patient First Visit (FPFV).19 In the survey, release of the EDC system after enrollment had begun was associated with significantly longer data entry time and the time from Last Patient Last Visit (LPLV) to database lock.19 Further, “always” releasing the EDC after FPFV was associated with data management cycle time metrics nearly double those for companies reporting “never” doing so.19 Release of the EDC system post-enrollment gives away the advantage of using EDC.16,19 [V]
Having the processes and tools in place at study start are necessary but not sufficient for gaining benefit from EDC. The data have to be up-to-date to be of maximal value. Failure to manage and maintain a study in an up-to-date state is a common point of failure for data managers and organizations – as the time from the patient visit to entry of data increases, the advantage of EDC decreases and the risk increases (Figure 1). Thus, in EDC studies, processes should be in place to facilitate and manage close to real-time data acquisition and processing.23,24,32,33

Key Relationships Between Cycle-time Metrics and Benefits of EDC.
a) Keeping Data Current Keeping Data Current
Keeping data current requires active management. Active management entails more than identifying important metrics, setting performance expectations and action limits, and tooling to detect when and where action limits have been reached. Examples of action limits include how late is a late query, too many answers to a question, too many missing fields. Follow-through of prompt and consistent intervention is the all-important last step. In other words, processes and tools to assure that the operational design of a study is implemented with high fidelity are important, but without systematic surveillance and prompt response when action limits are reached processes will degrade. “Ironically, there is a major difference between a process that is presumed through inaction to be error-free and one that monitors mistakes. The so-called error-free process will often fail to note mistakes when they occur.”34
Active management for up-to-date data starts with setting expectations for data receipt and discrepancy resolution timeliness with sites and vendors during contracting. Paper studies commonly used cycle-time expectations in weeks; e.g., data should be submitted within one week of the visit and queries should be resolved within one week of their generation. In an early two study EDC pilot, Dimenas reported 69% and 54% of visits entered same or next day and 23% and 24% of queries resolved same or next day.31 The average time from query generation to resolution of 18 and 17 days respectively for the two pilot studies.31 Last Patient Last Visit (LPLV) to clean file was 14 and 20 days respectively.31 In a cluster randomized experiment reported by Litchfield, et al. where investigational sites were randomized to data collection via EDC or paper, the majority of EDC data was entered within a few days after the visit with 90% of the data entered within three weeks after a study visit. This was a stark contrast to the paper group where data entry took up to six months.18 Timely data response can be incentivized through payment by clean visit or batch, often called milestone-based payments. Setting performance expectations is only the start of active management.
Active management of data acquisition and processing requires three things: (1) real-time or daily reporting of status and late items such as late entry or outstanding query responses in a format that facilitates follow-up as demonstrated in Figure 2 (an additional example is provided in Mitchel, et al.)32; (2) assigning responsibility to follow-up with each site and “work the report”; and (3) regular communication to leadership regarding the status. The latter provides an escalation path and leverage for getting the data in, in addition to the payment incentives. In this manner, expectations, incentives, and enforcement are aligned and work together to maintain up-to-date data entry and processing. Reporting late items and using the reports to drive action is called management by exception and is a critical aspect of active management. Metadata available in most EDC systems such as visit date, data entry time-stamp, query generation and resolution time-stamps, and data review time stamps can be used to support active management, hasten data processing activities, and ultimately detect problems and opportunities for improvement more quickly.
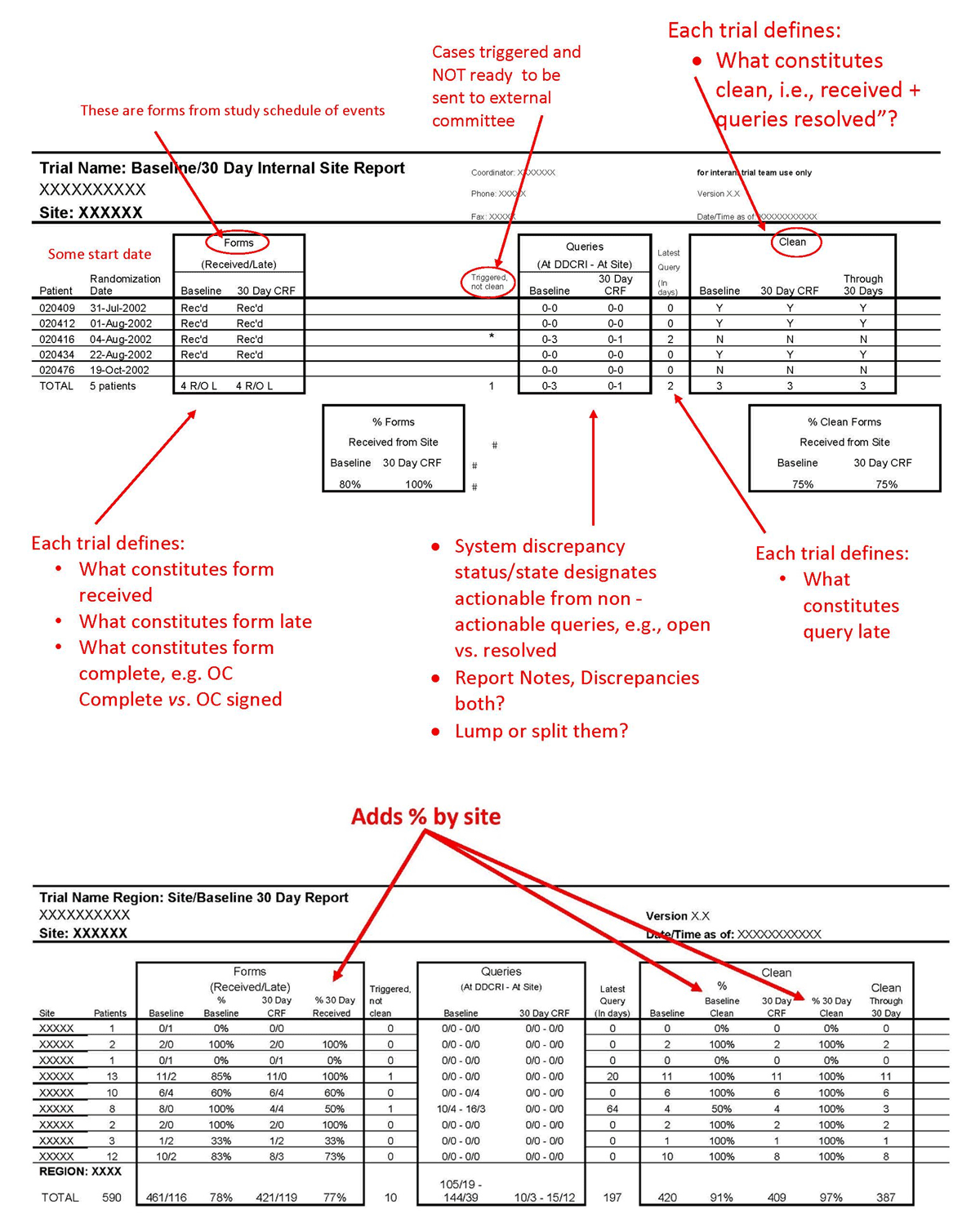
Reporting to Facilitate Late Entry/Resolution of Queries.
While some standard system status reports list only counts of cumulative or by-period completed items, others facilitate the work. For example, a CRF page status report that provides the number of complete and incomplete CRFs brings visibility to the data entry status. Doing so and following-up on outstanding pages as patient enrollment progresses avoids a large number of incomplete CRF page counts. The same can be done for any data collected for a study and for managing resolution of data discrepancies as well. Many EDC systems are able to link a user directly to the outstanding or late item so that the resolution of data discrepancies can be completed using the report as a work list to address the outstanding items, working sequentially down the list. Such directly actionable reports and within-system connectivity facilitates completion of work by eliminating steps. Directionally actionable work list reports should be available for all units of work such as visits or procedures, data from such collected via eCRFs, central labs, devices, or reading centers, as well as data discrepancies. Others have emphasized the importance of patient-level, site-level, and study-level reporting to actively manage data collection and processing.16,22,31 Applying active management by using an incremental form or casebook lock strategy to lock-as-you-go brings the arc of each subject visit to completion as soon as possible and reports start to show “clean data” soon after the study starts.23,24,33
An insightful industry leader mined data from over 100 EDC studies conducted by his organization.17 From experience and the data, three modes of data transmission and review behavior were identified (see Figure 3.) The number of days until the peak is the number of days that the organization could have had to identify and correct problems. Similar to outbreak response in public health, early detection and intervention prevent those that come after from experiencing the effect of the problem.
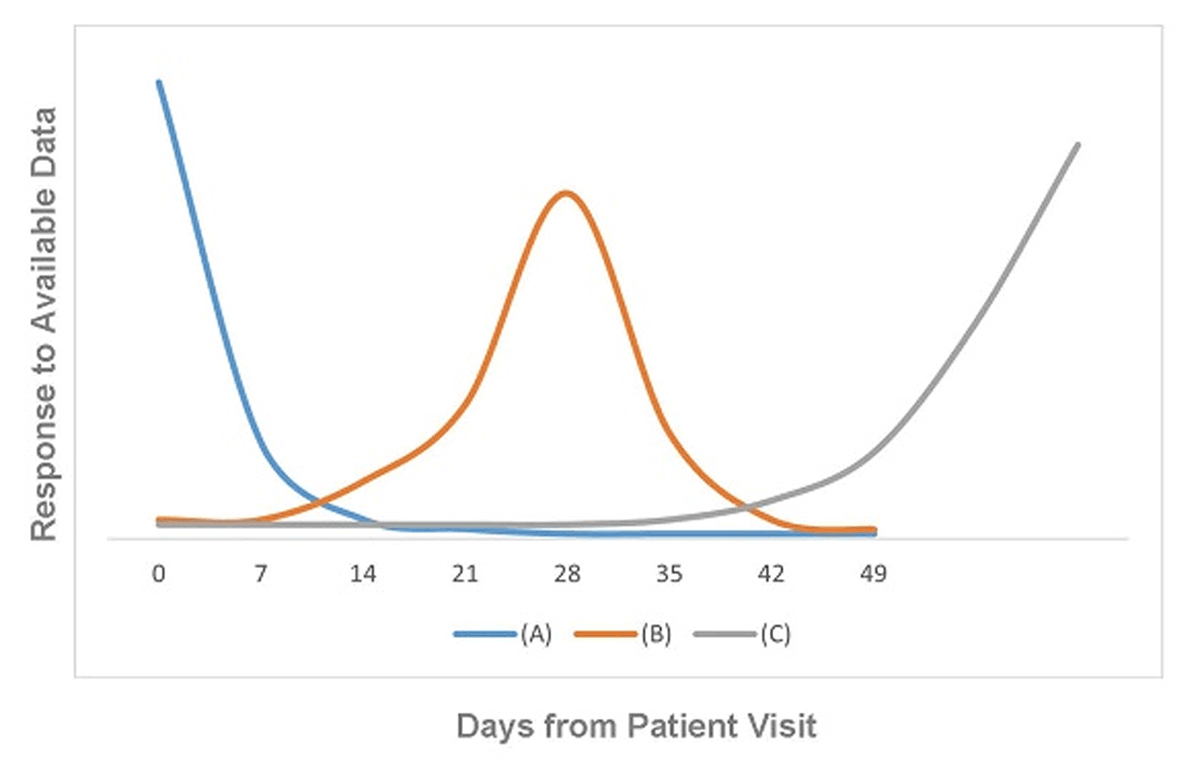
Three Patterns of Data Responsiveness. Adapted from Summa 2004.17
a) Ideal data responsiveness behavior. The majority of data are entered, resolved, or otherwise responded to at the earliest possible time. The peak should be highest at the earliest time point (zero days from the prior workflow step), should taper off precipitously, and the tail should be small. Further improvement in this ideal scenario is pursued through root cause analysis (understanding the root causes) for data entry, resolution, or response occurring later in time. Curves can be displayed by study, site, study coordinator, data manager, monitor, or any other actionable unit of analysis to help identify opportunities for improvement.
b) Likely systematic delay in responsiveness to the availability of data. The majority of data activity of interest (data entry in the figure) does not take place till around 14 days after the patient visit. This behavior is indicative of responding to the data just before some deadline. Importantly, data displayed on the graphs should be homogeneous with respect to deadlines; where deadlines are different for different sites, regions of the world, or in-house versus contract monitors, the differences in expectations will obscure the signal.
c) Waiting till the end. The majority of the data are responded to at the end of the period of interest.
The behavior in graph (A) in blue takes full advantage of immediate availability of data possible with EDC. The behavior in graph (B) in orange has room for improvement. The behavior in graph (C) squanders the opportunity for early detection and correction of problems possible with EDC.
“With EDC the batch processing has been replaced by the continuous flow of data.”17 With a continuous flow of data, active management and active surveillance become extremely useful. Older data review and trend analysis processes in paper-based studies were based on data that often lagged in time by a month or more with up to six months lag reported.17 In an EDC study however, site staff can enter data and view and resolve edit checks during or immediately after the subject’s visit. Data are quickly available for active management and surveillance processes. Delays with EDC processes are due to lack of active management rather than technology and, as such, persist on studies or in organizations lacking an active management culture and tools.17 Figure 4 approximates results achieved in a controlled experiment study comparing EDC to traditional paper data collection.18 Faster data availability, i.e., the space between the curves, offers up the ability to detect and resolve problems sooner and trigger risk-based action sooner than otherwise possible. The combination of EDC technology, active management, and active surveillance provides current data for decision-making and enables faster detection of problems. Failure to implement effective active management wastes this key advantage.17
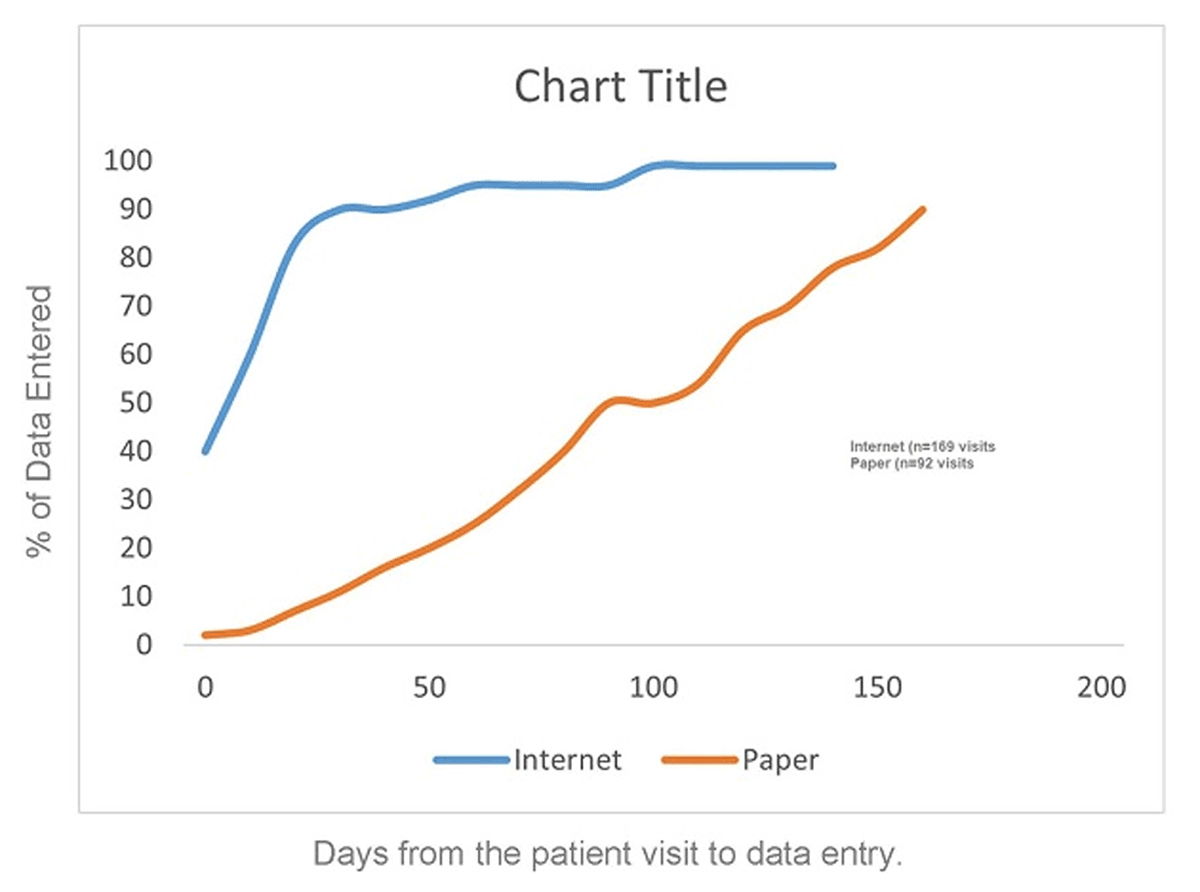
Percentage of Patients With Data Entered At Each Day Following the Visit.
Active management strategies have demonstrated through elapsed time metrics such as time from visit to entry and time from visit to clean data20 rapid detection of safety issues.23 In a subsequent study conducted by the same organization, 92% of data were entered within two days and 98% within eight days. In the same study 50% of the data were reviewed within 13 hours of data entry.24 For the same study, 22% of all queries inclusive of those generated by edit checks at the time of entry, nightly checks in the EDC system, and manual review by clinical trial monitors were resolved on the same day they were generated, 78% within five calendar days, 91% within ten days, and 99% within thirty days.24 Similar results were obtained in a comparison of six direct data entry studies to a traditional EDC study. In four of the six studies, 90% or more of data were entered on the day of the office visit, 88% of data were entered on the day of the office visit in the fifth study, and 62% of data were entered on the day of the office visit in the more complex oncology study. Within five days of the office visit, 75% of data were entered for the more complex oncology study and 82% within 10 days.33 Notably, two of the sites in the more complex oncology trial exhibited data entry time metrics comparable to those in the leading five studies.33 These results were achieved within the context of studies using Direct Data Entry (DDE), i.e., eSource entry during the study visit with transmission to a secure site controlled eSource data store and subsequent transmission to the EDC system for eSource data elements.32 These results are likely most easily achieved in that context where sites select eSource data elements, are trained to enter data for them immediately, and are managed to those expectations. DDE or not however, the principle of getting data into the study database as soon as possible after the visit and using the data to inform study management decisions is directly applicable to EDC. In contrast, to the entry times for the five DDE studies, the historical comparator EDC study where paper source was used had 39% of the data entered on the day of the office visit, and 75% of the data were entered within 10 days.33 In addition to achieving these data acquisition metrics, active management has demonstrated the ability to make faster, mid-course corrections to both protocols and EDC systems.24
b) Active Data Surveillance
Complete data management equally includes systematic screening to detect unexpected problems.34 The goal is to identify problems in study conduct, site operations, and data collection. Detecting unanticipated problems will never find all of the problems, but almost always identifies some. Possibly more importantly, broad systematic screening demonstrated a commitment to assuring subject safety and implementation fidelity of the protocol. Examples of protocol and operational events for such screening include the following.
Operational metrics examples:
-
Data submission and query response timeliness24,31 such as
visit date to data entered
data entered to data queried
data queried to query answered
query answered to query resolved
patient out to patient record locked
counts of automated queries to detect over-firing or under-firing rules
number of queries per patient31
frequency of queries by field to detect problems with the eCRF
CRA review status of the eCRF forms24
-
Metrics for data changes24 such as
help desk calls, tickets, or error reports
system and support response times specified in the Service Level Agreement21
system downtime
Protocol metrics examples:
Percentages of subjects meeting key inclusion and exclusion criteria
Protocol violation percentages
Elapsed time windows for visits and study procedures
Differences in distributional characteristics of data values
Percentage of subjects with use of concomitant or rescue medications
Percentage of subjects with reported adverse events
Percentages, proportions, or rates of early terminations and reasons for termination
Many metrics of interest are aggregates of different aspects of units of work, for example, average data entry latency, query response times, number of queries per form, and percent of time points for which ePRO data are missing. Additionally reported adverse event patient visit should be available and run early in a trial to identify potential problems. Remediation can then be taken to reduce or even eliminate underlying problems as the study progresses. Remediation may include revisions to CRF forms or completion guidelines, retraining site staff, retraining CDM staff regarding query wording, adding alerts or other supportive workflow, or reiterating expectations through special topic newsletters and teleconferences.
All of these allow comparison of data across sites to identify unwanted process differences at sites. In fact, one of the newer CDM competencies is, “Applies analytics to identify data and operational problems and opportunities.”35
Such screening requires making risk-based and potentially statistically-based decisions about how different is different enough to look into, and the extent of investigation and attempts at resolution. Examples of resolution may include retraining the site concerning the protocol, the EDC system, eCRF, or eCRF completion guidelines; explaining problems to sites in an auto-notification message, reminder, or banner via the EDC communication functionality; or reviewing top problems and corrective action in periodic meetings and presentations to sites. Similarly, screening query frequencies may prompt the project team to re-examine edit check specifications and narrow or broaden ranges, change logic, or eliminate certain checks altogether.31
The sequence and timing of initiating surveillance must be decided early. Data are usually entered into an EDC system before they have been source reviewed. Data validation and review activities are usually performed before data are source verified, if the latter is planned for the study. In a Risk-Based Monitoring (RBM) paradigm, results of surveillance can be used to trigger or prioritize monitoring calls, visits, or SDV. To the extent that data cleaning, active surveillance, and SDV are intertwined, communication and workflow between clinical research associates (CRAs) and CDMs should be established within the EDC system. For example, many EDC systems have the functionality to trigger SDV or to track SDV and indicate completion by form, visit, or subject.38
The Mitchel, et al. report details their approach to implementing RBM. Briefly, the trial team identified risks to patient safety and the trial results and scored the likelihood and severity of each.24 They then developed reports and alerts for early detection as well as a plan to target these risks that included review of source documents, schedule for on-site monitoring, monitoring tasks to assure protocol and regulatory compliance, frequency for central monitoring, and documenting monitoring in the EDC system. Active management was a pillar of the approach and the authors concluded that time to using the data, e.g., in surveillance to detect a risk event or data review by monitors, was a key factor in realizing RBM.24
Operationalizing active surveillance requires CDM such as
encoding them in written procedures
developing tools to screen data and report the results
dedicating resources to manage the process and work the reports
development and delivery of role-based training for the study team on those processes and tools
Many EDC systems have existing reports for some of these. However, today, not all data are integrated within the EDC system. Some data are managed in other systems or organizations. Comprehensive data surveillance as described above requires timely access to all of the study data; thus, obtaining the full benefit from EDC would be best with interoperability and data integration with all data sources. Metric reports from other data sources should also be considered. Studies integrating data from automated equipment such as an electrocardiogram (ECG), personal digital assistant (PDA), Interactive Voice/Web Response System (IVRS/IWRS), or other electronic devices for electronic patient-reported outcomes (ePRO) often necessitates development of custom reports.
Application of advanced statistical techniques to filter and detect more significant anomalies often necessitate use of a statistical analysis software package such as SAS® or R®. Such ongoing surveillance has also been reported when new systems or new functions were implemented to ensure that the EDC system continues to operate as intended.22
c) Job aids for the sites
Variability in site-specific practices is inevitable. However, site-specific procedures such as insertion of additional data processing steps can interfere with the advantages of EDC. Reports of sites using paper CRF-like forms or worksheets as source or as an intermediate step on which to record data as it is abstracted from the source and from which data are subsequently entered into the EDC system have been reported in the literature.27,31,37,38 This practice doubled the data entry workload at the sites.31 In some cases, the temptation exists to assign the data entry of the worksheets to staff who are not familiar with the patients and their clinical course. Doing so invites additional error by use of single entry by staff less familiar with the data and should be discouraged.37 [VI] In addition, every transcription process adds opportunity for error37 and also increases the monitor’s workload because, at least for critical variables, the paper form worksheet and eCRF need to be proofread and compared unless those data elements as entered into the EDC data are SDV’d.2,31,37
7) Communication Plan
Many EDC systems offer functionality to support communication between the central study team members and site personnel. Where possible and practical, such system functionality should be leveraged for communication about trial conduct tasks. Computers perform such deterministic and repetitive tasks with higher reliability than humans and without the delay of waiting for humans to read and respond to email. For example, flagging discrepant data in the EDC system, by applying a workflow for discrepancy management, handles the “communication” regarding discrepancy resolution through the system as a by-product of the different roles undertaking actions toward discrepancy identification and resolution and obviates the need for emails to request responses or notify that resolutions have been completed. Likewise for other workflows such as safety event notification and tracking and prompting collection of source documents for Clinical Event Classification (CEC) review.
Provision of trial information and job aids to clinical investigational sites is also an important type of communication and can be managed through portal functionality in many EDC systems. Similarly, many EDC systems are able to provide status and other reports within the system. Doing so at a regular frequency or real-time obviates email distribution, drives site personnel and central team members to the EDC system where addressing outstanding issues may be only a few clicks away, and promotes the EDC system as the source of up-to-date information.
When it is not possible or practical to leverage the EDC system for needed communication, traditional communication vehicles such as calls, meetings, and email are needed. The purpose, frequency, distribution, and content of each should be considered and documented.
8) Security
Privacy regulation and guidance, such as the HIPAA privacy rule, ICH Guidelines E6 Sections 2.11, and 4.8.10, and Article 8 of EU Directive 95/46/EC generally expect that access to an EDC system should be limited to authorized staff.2,39,40 Further, Title 21 CFR Part 11 requires the same for the purpose of ensuring that electronic records and signatures are as trustworthy as those maintained on paper.3 Controlling system access is part of system security, as are measures to prevent unauthorized access, adulteration, or use of study data. Such measures include required authentication of users, seclusion of information systems behind firewalls, and other methods of preventing external access, and programs for detecting intrusion attempts. While the technology and methods for ensuring information system security are beyond the scope of this chapter, controlling user access and privileges within the system is often part of CDM’s responsibility.
Security cannot be achieved without user compliance. As such, site user training is often provided by data managers as part of study start-up as described in the GCDMP chapter titled EDC – Implementation and Study Start-up. During training all users should be informed about system access rules. During monitoring visits, the sponsor or designee should remind site staff of the importance of confidentiality for each user’s ID and password and should be vigilant for signs of security weaknesses such as user identifiers or passwords posted in plain sight or sharing login credentials. Suspected noncompliance with access rules should be reported to the assigned system administrator as appropriate.
a) Keeping Data Current Maintaining System Rights Determined by Roles and Privacy
EDC systems generally use role-based security, where different system actions can be performed or accessed for specific user roles. System privileges such as entering or updating data, adding a new patient, responding to queries, or performing SDV are assigned to roles. When users are granted system access, usually through provision of login credentials, they are usually also assigned to a role in the system. The role then confers privileges for what can and cannot be accomplished within the system by the user. Study-specific role and responsibility definitions and corresponding user access and privileges are often maintained in a matrix showing privileges corresponding to each role. Documentation of system access must comprehensively represent all system access throughout the study.3 [I] Such documentation assists auditors with understanding who had what type of access over what time period. Throughout the course of a trial, roles and responsibilities may change, as may CDM processes (for example, a monitor may no longer be allowed to close queries.) Because these may impact system access or privileges, any changes to documentation describing access rights should be tracked.3 [I] Such changes should be communicated to all study team members. [VI]
b) Managing Periodic System Access Review
Managing user accounts and permissions is a time-consuming task, requiring diligence to ensure security and confidentiality are maintained throughout the duration of a trial. Open communication with clinical operations is necessary to keep track of site and contract research organization (CRO) staff changes so as to activate or deactivate corresponding user accounts as needed. User access to the EDC system and all other study data should be periodically reviewed.3 [I] Additionally, as part of this periodic review, appropriateness of access rights for each user should be verified. [VI] The review frequency depends on study and organizational factors and should be determined by organizational procedures.3 [I]
c) Managing Conventions for User Login IDs and Passwords
Each user of an EDC system should have an individual account, consisting of unique credentials, often a login ID and password. Typically, the initial login ID and password can be sent to the individual user using his or her e-mail address, or through traditional methods such as mail or courier. The system administrator should only grant a user access to the system once the user’s role-specific training has been completed and documented. [VI]
If credentials were supplied to a user or otherwise known by others, the user should be required to change their initial login ID and/or password when a user first logs into the EDC system.3 [I] If the system is not capable of forcing the user to change their password on first entry, trainers will need to ensure this activity is discussed with all trainees. Users should be trained to keep their IDs and passwords confidential.3 [I] Each login ID should uniquely identify the user within the EDC system’s audit trail, and enable tracking of any information that the user enters, modifies, or deletes. Additionally, users should be instructed to log onto their account, complete data entry and review, and log out at the completion of review.3 [I] Users should be instructed to log out of the EDC system when the personal computer (PC) used to access the EDC system is left unattended.3 [I] Login ID and password requirements should include restrictions on re-use of accounts and passwords, minimum length of login IDs and passwords, required frequency of password changes, and automatic log-off when a PC accessing the EDC system exceeds a predetermined amount of inactive time.3 [I]
d) Managing User Access
Turnover of site and study team members is likely. Therefore, management of user access will be an ongoing task throughout the course of an EDC study. Procedures should be in place for revoking access when users change roles or leave the project or organization.3 [I] Likewise, procedures should cover granting accounts to new users. Monitoring user access will likely require both CDM and clinical operations resources to manage site and sponsor user access.
Disabling Access During a Study
Procedures should be established to define processes for disabling or revoking access to the system as needed.3 [I] These processes should clearly define who is responsible for communicating staff changes (both internal and external), documenting these changes, and executing these changes. [VI] Requirements for automatic deactivation of accounts should also be established in the event of security breaches or users who do not log in for extended periods, such as not accessing the study within 90 days or some other specified time frame. [VI]
The sponsor should define appropriate lock-out rules in the event of unauthorized access, whether attempted or successful. [VI] If a user enters an incorrect ID or password, an alternative method, as specified through established standard operating procedures (SOPs) or work instructions, should be employed to quickly reauthenticate the user and to quickly reestablish system access for reauthenticated users. [VI]
Adding New Access During a Study
Throughout the course of a trial, it will become necessary to add new users or modify access privileges for existing users. Procedures should be established to ensure these tasks occur without disruption of ongoing study activities. [VI] These procedures should detail training prerequisites, steps for requesting access, and the staff members who are responsible for ensuring all site staff and study team members have appropriate access. [VI] Documentation of completed training should be provided to the appropriate personnel so they know which users may be granted new or modified access rights. [VI] Documentation should be maintained throughout the course of the study and archived with study documentation.3 [I]
9) Ensuring Effective Software Support
When available, reports (which may include surveys) detailing the responsiveness and effectiveness of software support (e.g., the average length of time the help desk takes to assist a user) should be reviewed regularly to ensure support is effective. Several factors are important to ensure assistance is provided efficiently and expeditiously, including easy access to support staff, ability to address users’ questions, and the availability of support when needed.
a) Keeping Data Current Providing Multiple Language Support
Although language needs for the help desk should be determined during the pre-production phase of a study, CDM staff should be sensitive to complaints regarding communication problems during the study conduct phase. [VI] The problems may be, in part or in whole, related to an inability of the help desk to provide the language support needed, and may require a revision to the original translation needs of the study.
b) Providing 24 × 7 × 365 Support
As with multiple language support, help desk availability should be determined prior to the start of a study. However, during the conduct of the study CDM should evaluate feedback from users to ensure that the availability of support is adequate for the study. [VI] Reports detailing the responsiveness and effectiveness of software support should be reviewed regularly to ensure that front-line software support is effective. [VI] Front-line software support is the lowest level of support needed and includes activities such as unlocking user accounts and resetting user passwords. Information gained from reports and feedback may involve reevaluating the original decisions regarding the level of support needed. For example, if 24 × 7 × 365 support was not originally set up, it may be necessary to reconsider the cost.
c) Down-time procedures
Where the EDC technology is supporting subject safety or regulatory requirements, down-time procedures for operational continuity when the system is down as well as back-up and recovery procedures must be in place.21 System users should be trained on down-time procedures and such procedures should be redundantly available through a mechanism other than the EDC system. [VI] “And for those non-emergencies when the site experiences interrupted internet access, the site can collect data on paper, enter the data when the internet is available and create electronic certified copies of the paper records.”23
10) Training
EDC-related training should be provided to anyone who uses the system. [VI] Training is most effective when provided as close as possible to the time when the newly learned skills will be used. If a significant time lapse occurs between training and use of the learned skills, retraining should be considered.
a) Reviewing and Maintaining Training Materials
EDC system training is an important part of proper study management. Training is dependent on the study and target audience; therefore, training materials should be developed with these considerations in mind to make the training as effective and appropriate as possible. [VI] Moreover, training should be an ongoing process, not just a one-time event. [VI] An EDC system can provide the sponsor with the ability to identify a need for retraining users. Some EDC systems can also be used by the study team to deliver updated training materials and communications to users in a timely manner. For example, updated CRF instructions can be immediately provided to all sites and study team members, and newsletters can be provided through a dedicated website to communicate updates or changes.
Identifying users’ needs for retraining is an important activity of both CDM and clinical operations team members who interact with the site regularly. A mechanism should be in place for the CDM to become aware of situations at a site that may present challenges and a need for retraining, such as coordinator inexperience, isolation, turnover, or competing priorities. [VI] Available information, such as help desk reports, query frequency reports, and protocol deviation reports, can be used to identify materials that need to be updated or users requiring new or additional training. For example, with query frequency reports, one site can have a misunderstanding of how specific data is entered into the system and one site can have a misunderstanding of what specific data is entered into the system. The report helps identify a need for site or system training.
b) Ensuring Site and Sponsor Staff Training During Turnover
A common occurrence in clinical research is turnover of both site and sponsor staff. New staff should receive required training. [VI] A plan should be established for new users to be trained in a timely manner so they will have the benefit of access to data on the EDC system. [VI] If new site staff are not trained and do not have access to the system, they cannot enter data, and study timelines can be negatively affected. To ensure regulatory compliance, controls should be put into place to ensure untrained users do not have access to the system.3 [I]
11) Reporting
a) Mid-Study Requests for Subject Data
A mid-study request for subject data can occur for many reasons, including, but not limited to
A scheduled interim statistical analysis based on study design and protocol, which typically focuses on efficacy data
An interim review of data focusing on safety data, such as adverse events and other data that indicated safety issues in earlier studies (e.g., ECG data, lab panels)
DSMB or Clinical Endpoint Committee (CEC) regularly scheduled meetings
A submission package or other type of update (e.g., 120-day safety update) for regulatory purposes
Any other planned or unplanned data lock
A major factor affecting delivery of mid-study subject data is whether the data are stored by the sponsor or a vendor. If data are stored by the sponsor, the data should be readily available, thereby reducing costs and resources needed. If a vendor’s hosted system (Application Service Provider (ASP) model) is used, the timing and frequency of deliveries are more important.
Whether a sponsor or vendor system is used, the required subject data should be clearly identified. [VI] Examples of prerequisite identification for exporting subject data include but are not limited to.
An interim analysis planned to occur at a particular milestone (e.g., the 100th randomized patient) or of a particular module (e.g., dosing data)
A safety review planned to occur at a particular milestone (e.g., 25% patients enrolled, 50% enrolled)
A mid-study efficacy analysis based on statistical design of the protocol
Regularly scheduled DSMB/CEC meeting
In addition to determining which subjects are to be included in an export, the sponsor should identify which records are to be included in the delivery. [VI] The simplest solution is to include all study data, regardless of its status. However, delivery could be restricted to data verified by the CRA or monitor, or to locked (clean) data, which requires close coordination with the CRA for scheduling monitoring visits. If data are to be used for an interim safety analysis, reconciliation of SAEs and Medical Coding may require additional attention.
Any external data that is to be integrated into the database prior to providing any subject data mid-study (e.g., laboratory data or ECGs) should be planned in advance of the study team’s timeline for reporting. [VI] As necessary, the completeness and accuracy of such data should be ensured by reconciliation before the data delivery occurs. [VI]
The recipients of requested study data and the impact to study blinding should also be considered. [VI] For interim analyses, datasets are typically provided to a biostatistician or statistical programmer, who subsequently creates tables or listings from that data. Timing of the delivery (e.g., planned or on demand) is also an important component to consider. If required data deliveries are scheduled, necessary procedures can be planned in detail. [VI] However, if ad hoc requests for data are anticipated, the process for exporting and delivering data should be defined in the SOPs or Data Management Plan. [VI] When ad hoc requests are received, programs should be tested and validated to ensure timely delivery.3 [I] Testing should include the complete extraction and delivery process, including checking that all required variables are available in the datasets and populated with expected values. [VI] Errors or omissions noted during testing should be corrected until the data export operates as required.
b) Mid-Study Requests for Notable Subject CRFs
Regulatory agencies such as the Food and Drug Administration (FDA) require CRFs from subjects to meet certain criteria. As required by CFR 314.50(f), for any new drug application (NDA), individual CRFs are to be provided for any subject who withdrew from the study due to an adverse event, or who died during the study.41 [I] Depending on the study and FDA center, the FDA may request additional CRFs for review of the NDA.
The sponsor should be prepared to transfer CRFs at any time during the study such as for an NDA periodic safety update or integrated safety summary. One possible solution is to provide electronic copies of CRF images. When working with a vendor, the sponsor should factor the process for obtaining CRFs in the contract’s study timelines and expectations (e.g., maximum number of requests).
12) Measuring and Controlling Data Quality with EDC
Web-based EDC brings unique data quality measurement challenges. The process of getting data into the EDC system may be handled differently at clinical sites. Some sites will use paper CRF-like worksheets on which to initially transcribe data abstracted from source documents such as the medical record. The paper form will then subsequently be entered into the EDC system. Other sites will abstract the needed data and enter it directly into the EDC system. In both cases there is a source document. The difference in the two approaches is the use (or not) of the paper form as an intermediate step. Either way, the error rate between the source and the EDC system (1) is needed (per ICH E6 R2 ss 5.1.3) and (2) can only be obtained by SDV.2,38 Keeping records of the number of data values SDV’d and the number of discrepancies detected has historically not been a consistent monitoring responsibility.
With the transition to web-based EDC, this additional task necessary to measure the error rate was not routinely added to monitoring responsibilities; thus, few organizations measure the source to EDC error rate.37 Historically, with EDC the number of queries has been used as a surrogate to indicate data quality; i.e., the more discrepancies detected by the query rules, the lower the presumed quality.18,20,42,43,44 As noted by Zozus et al., medical record abstraction, i.e., the manual review of medical records, selection of data need for a study, and transcribing onto or into a study data collection form, electronic or otherwise, is associated with error rates an order of magnitude higher than other data collection and processing methods.45 Thus, the source-to-EDC error rate should be measured. [III] Such measurement will meet the requirements in ICH E6 (R2) section 5.1.3 if it is measured from the source to the EDC system or if the steps from the source to the EDC system are covered by review such as a comparison from the analysis datasets to the source. [VI]
Further, use of EDC does not change the fact that medical record abstraction is a manual process. As such, ongoing measurement of the error rate and feedback to sites is strongly recommended to maintain the error rate within acceptable limits for the study.46 (Zozus 2015) [III] As noted by Helms, in the case where the EDC is used as the source, i.e., EDC eSource, obtaining an error rate requires an independent source of the same information, similar to use of split samples sent to two different labs.37
The EDC data audit process may differ between organizations. The auditing process will be impacted by how the data can be extracted from the EDC system and the source used for comparison. Any additional programming required to transform study data into SAS® datasets could affect how data are displayed. Additional EDC issues to consider for auditing include, but are not limited to, reconciling medical coding, data management plan comparison, external data import, the extent of process auditing, query verification, and completion of all queries that required data changes.
An audit plan should be established in advance to identify approaches to be taken, including sampling, establishing sources for comparison, acceptable error rates, how audit findings will be reported, and expectations for corrective and preventative action.
a) Keeping Data Current Change Control
Any EDC system may undergo changes during the conduct of a study because of changes in EDC software and/or changes in the study itself. Efficient change control processes are needed to quickly address issues that arise during a study to prevent further occurrences and to implement corrective and preventative action with minimal disruption.21 [VI, VII] Though speed is important, changes should be fully explored to minimize adverse impact on existing data.21 [VI, VII]
b) System Change Control
Because clinical trials may occur over the course of several years, software changes and upgrades will inevitably have an impact on EDC studies. These changes or upgrades are not just limited to core EDC software, but could also include upgrades to the operating system, back-end database software, or any auxiliary software integrated with the EDC system, such as reporting or extracting software. The differences in change control strategies and processes depend on whether the system is developed internally by the sponsor or purchased from a vendor.
c) System Validation
If software was purchased, the sponsor may decide to rely on the vendor’s system validation package for the software, including all releases or upgrades, and maintain the system as a “qualified” platform rather than performing system validation upon each release. [VI] However, “qualified” software platforms should not be customized by the sponsor unless validation of the customized platform will also be performed.3 [I]
d) Controlling Changes to the System by Incorporating Software Development Life Cycle Principles
In order to implement upgrades to the software system (whether it is a new release, a minor version update or a SAS® update), CDM representatives should make a complete assessment of the software changes and obtain input from other functional areas that may be impacted, including a thorough risk assessment. [VI] Documentation of such should be maintained for each version release.3 [I]
e) Risk Benefit of Validation Efforts
The first step in performing an assessment is to gain a clear understanding of all changes or additions that will be made to the software. For software purchased from a vendor, this task can be accomplished by ensuring that the software release notes are reviewed and well understood by appropriate staff. Release notes should include documentation of all changes, any known issues in the new release, and instructions for upgrading the software from previous versions.
For software produced internally by the sponsor, a well-developed change control process should be established. This process should include steps for reviewing change requests, grouping multiple change requests together as appropriate, updating requirements and design documentation, build, testing, and implementation.
To determine whether a software system should be upgraded, the sponsor should consider the following issues:
Impact on data – Assess if any changes in software functionality could potentially impact data integrity. For example, if certain characters or functions will no longer be supported, the sponsor must make sure data integrity will be preserved after the software upgrade. [VI]
Impact on existing code – The software upgrade may require you to make changes to existing programming code. [VI]
Auxiliary systems – The sponsor should assess how related systems or programs will be affected by the software upgrade. Will other systems require corresponding upgrades or modifications? [VI]
Impact on sites – Will the study be inaccessible during the software upgrade? Is the site required to perform certain tasks such as installing software on their local PCs or changing browser settings? Will the site require additional training? How will the sites be notified of the impact? [VI]
Comparison of cost and value – The costs of implementing and validating a software upgrade should be compared with the business value to be gained. [VI]
The impact on ongoing studies – Considering the impact on the study database and remaining duration, is it worth upgrading software to a new version? Does the software for ongoing studies need to be upgraded simultaneously? [VI]
SOPs and training materials – Will the software upgrade require revision of the sponsor’s SOPs or training materials? [VI]
For internally produced or customized EDC software, new requirements documentation should be created.3 [I] This effort is often led by clinical data management. The requirements documentation should include new features and functionality, as well as changes to current features and functionality.3 [I] The requirements documentation serves as the basis for design specifications. Creating design specifications is typically performed by the group who will be programming the changes.
In addition to the requirements documentation, clinical data management will need to develop a test strategy that documents the testing and validation required for the new software. Depending on the type of upgrade, intensive testing is not always necessary. The following guidelines can be used to determine required testing efforts:
For a minor version (bug fix or upgrade), limited testing may suffice. [VI]
For a new release or major version upgrade, moderate to intensive testing is usually advisable. [VI]
For purchased EDC systems, the vendor should be able to provide and maintain testing plans and results. [VI] For internally produced EDC software, test scripts or test cases based on new user requirements should be produced.3 [I] Before implementing changes in a production environment, all testing should be performed in a test environment. [VI] New features and functionality provided by the upgrade (as well as enhancements of existing features or functionality) should be tested.3 [I] A problem log or Web-based error tracking system should be employed to track errors found during testing, so the status of these issues can be monitored through to their resolution.3 [I]
After validation of the new release has been successfully completed, the new version or changes can be implemented in production. Please refer to the “Database Validation, Programming and Standards” chapter of Good Clinical Data Management Practices for more information about validation, including recommendations, minimum standards, and best practices.
f) Training on Changed Software
While a minor upgrade to software is likely to go unnoticed by users, a new release or major upgrade to software could require additional training. The sponsor should determine the required level of training, which users should receive training, and the method of providing the training. [VI]
Typically, sponsor staff (CRAs/CDM) and site staff will require training, which can be delivered in person, from durable media, or over the internet. Presentations using screen images can be particularly beneficial for training purposes, as they can be reused for later training sessions. Sponsor staff should be trained first on the software’s new or modified functionality and then the site staff. [VI]
g) Developing the Rollout Plan
Before making new software available to staff, the impact of the revised software needs to be assessed. [VI] For example, if software revisions will require modification of approved CRFs, the sponsor should identify Institutional Review Board (IRB) issues to be addressed (IRB issues are not likely to apply to a software upgrade but may apply to CRF revisions). [VI] The sponsor should determine whether new software should be upgraded in stages or all at one time.
Sites should be informed of the rollout of new software with sufficient time given for necessary preparations. In case the upgrade does not occur as expected, a clearly defined rollback or fallback plan should be established prior to software implementation. [VI] For international studies, the time available to perform software upgrades can be limited and may require the upgrade to be completed during normal business hours.
h) Managing the Legacy Release
Software vendors typically maintain all software versions for a defined period of time. The sponsor should be aware of the level of support provided by these vendors. When a vendor rolls out a new system, they may not continue to offer the same level of support for earlier versions of the software system and may eventually retire earlier versions. Typically, once a version is retired, the software vendor no longer provides support for that version.
Because of ongoing development of software systems, the sponsor should plan for future changes, and determine when it is appropriate to upgrade or retire an existing system. Some factors to consider include
Software upgraded during study conduct
Vendor’s support commitment to previous versions of software
Software or hardware that becomes obsolete
Decreased system performance
Trial timelines
i) Application/Study-Specific Change Control – Mid-study Protocol Updates
In contrast to software changes, a trial could also be affected by study-specific changes, such as protocol amendments or development issues. As a result, CRFs, edit checks, and/or reports may need to be modified. As change control is a heavily regulated process, any change should be documented, analyzed, and tested.3 [I] CDM should assess required changes to determine how they should be implemented in the system and deployed to sites. [VI]
j) Changes to CRFs
Version-control software should be used for CRFs. [VI] Electronic files may be organized by study in a hierarchical directory structure. For each study, initial release of and subsequent changes to CRFs should be indicated, e.g., by directory naming conventions or labels. [VI] Identifying the date and time files were released must be possible so the release timeline is clear for regulatory purposes or to troubleshoot issues with CRFs.3 [I] A log documenting the version release(s) should also be maintained and signed by all appropriate parties as required and defined by sponsor procedures.
Before rolling out the new version of a CRF, CDM may need to assess who the changes will impact. If the changes resulted in revisions to already approved CRFs, the reviewer should determine if these changes will impact the sites and IRBs and inform all appropriate study team members and sites well in advance. [VI] For international studies, the time to deploy an updated version is more limited and may require deployment during normal business hours.
CDM should consult with clinical operations to determine whether to roll out the new version in stages or all at once. If changes are rolled out mid-study, the study team should first be notified when the changes will be available, and whether the study team will have an opportunity to review changes prior to deployment to sites. [VI] Site staff should be notified of training and rollout before changes are released to the production system. [VI] Once changes have been made, all parties should be notified. [VI] To ensure appropriate sites are moved to the new version of the CRF, CDM should create a log that will keep track of when each site was moved to the new CRF version. [VI] Proper logging will ensure no sites are missed in the process. Not all sites may require the new version, such as in cases where the changes are related to a protocol addendum. Target dates should be set and tracked for upgrading sites to the new system, which should be closely monitored and tracked. [VI]
Data entered in the previous version of the EDC study should be made available to the sites and study team.5 [I] Any new data that have not been entered into the previous version of the EDC study usually follow the newly released CRF format. If edit checks were modified, CDM should review old discrepancies to determine if they are still valid, and if any of the discrepancies need to be closed due to the changes. [VI]
13) Final Review
The final review precedes the database lock process. A list of all close-out activities and what triggers them should be used.21 [VI, VII]
a) Verifying Source Document Verification of All Records
Before a form can be locked and the database closed, source documentation and data review should be completed as required for the study for all forms and fields. [VI] It is beneficial if the EDC system can indicate SDV status and data review activity. Prior to locking a database, ensure all required SDV has been completed by the CRAs.
Frequent communication between the clinical study team, including the CRAs and clinical data management team, is critical. Changes to data in the CRFs are possible as late as the day of database lock. A plan should be established to ensure CRAs are available to source verify any changes, if necessary. [VI]
b) Verifying All Queries Have Been Closed
All reasonable efforts should be made to ensure all queries are answered or closed prior to database lock, particularly for those queries that may impact the analysis or outcome of study results. [VI] Depending on the details of the Data Management Plan (DMP) it may be acceptable to lock noncritical forms with open queries. During study startup, conditions for locking a CRF should have been defined thereby ensuring, for example, that the CRF cannot be locked with open query status. Prior to locking a database, CDM should ensure all queries have been answered or closed, including automatic queries and manual queries created by CRAs and clinical data managers. [VI] This task may be accomplished through use of varied system reports and status indicators, according to the EDC system’s features.
c) Verifying e-Signatures
It may be necessary to verify that all CRFs have been electronically signed by the investigators (principal and/or sub) responsible for a particular site. The investigators are responsible for reviewing each subject’s CRF to confirm that data entered are complete and accurate. Sponsor organizations must determine the level of detail required for investigators’ signatures. [VI] For example, some organizations will decide that the investigator’s e-signature must be applied to every individual CRF, while others may decide one investigator signature on the final page of each subject’s CRF is acceptable.
Ensure investigators and other site staff members are educated in the signature-break process long before study closeout so there is no confusion on the topic of closeout. [VI] Signature-break may occur when data has been changed post investigator signature. Should signature-break occur, this information will help avoid confusion, and will ensure the investigator is available to re-sign if necessary.
While there are many studies using e-Signatures, some are also still working with paper-based signatures, even with those studies utilizing EDC.
Regardless of the final signature method being used, a process should be established for notifying sites that CRFs are ready for investigator signature. Policies and processes related to re-signing a CRF should also be defined and adhered to. [VI] If a site changes a data point on a CRF already signed by the investigator, the rules must be in place to decide whether the data change “breaks” the signature. If the signature is broken due to the change, the investigator must re-sign the CRF.
It is expected that the CRA will track the investigator’s progress signing the CRFs. However, the clinical data manager may be responsible for conclusively verifying that all CRFs have been signed by the investigator prior to database lock. For any data changes that “break” the signature, the clinical data manager must verify those CRFs are re-signed. [VI]
14) Final Locking of Data and Database
The term “lock” may refer to not only locking a study or database, but may also refer to locking specific forms or casebooks. While not all topics discussed in this EDC-specific chapter actually occur during study closeout, they are common components of the database lock process and closeout of a study. For detailed information on a database lock, reference the GCDMP chapter on Database Closure. It is recommended that a checklist be established to ensure completion of required tasks prior to database lock.21 [VI, VII] As in final reviews, it is important that the planned activities are undertaken and occur systematically. As such, a list of all close-out activities, what triggers them, and how completion is achieved, including any criteria through which those decisions are made, is needed.21 [VI, VII]
Tasks on such a checklist may include, but are not limited to
Identifying personnel responsible for carrying out database close-out and lock activities
Ensuring all data have been received and outstanding data have been addressed.
Ensuring required medical coding of adverse events, prior and/or concomitant medications, and medical history verbatim terms has been completed; i.e., that all uncoded terms have been addressed and any quality review of the coding has been completed and the results are within established acceptance criteria.
Ensuring that all data discrepancies including those performed within and outside the EDC system and identified during data listing reviews have been resolved
Importing and/or reconciling all external data (and listing external data reconciled), for example, serious adverse event (SAE) reconciliation
Exceptions identified during final reviews and final checks sometimes are unanticipated, are not covered by pre-defined acceptance criteria, and require decisions be made and documented. Preparing for this eventuality by calling close-out meetings of decision-makers to review and decide such exceptions while data are still blinded (in the case of a blinded study) helps to keep close-out on track. [VI]
Once all database lock tasks have been completed, the database can be closed and soft- or hard-locked according to the sponsor-approved definition. This step implies that all forms in the study have been locked according to defined procedures, all tasks are completed, all conditions have been met as defined in the data management plan, and the final data transfer has been received or extracted according to data extraction specifications defined at the start of the study.
Planned QA and QC audits should be performed before database lock. [VI] Should an audit be performed on locked data? Based on findings from the audit of final data, further data corrections may be requested of the site. Once corrections have been made and verified, and additional investigator signatures have been obtained as necessary, another data transfer or extraction should occur. It is recommended that a comparison program be run to determine if requested changes to the database were successfully executed, as well as any other changes that were made to data other than what was expected. [VI]
a) Soft Database Lock
Typically, records that are soft-locked cannot be updated by sites, but the sites may still be able to respond to open queries. Many EDC systems support soft locks at a visit, page, or data point level. This capability supports a process of “rolling” or gradual soft locking of data throughout the course of a study, reducing the effort required by the CDM to lock data at the end of the study. Incremental soft locks can also be an effective approach to supporting mid-study or interim data review activities.
b) Hard Database Lock
Typically, a database hard lock occurs when all data have achieved soft lock status and all other study closeout activities have been completed. After a database has been hard-locked, tightly controlled procedures for unlocking the database must be employed, and only a few privileged users should be able to modify data. [VI] Once the database has undergone a hard lock, the data are considered ready for final analysis and archiving.
c) User or System Access Revocation
At the conclusion of a study, user access rights must be modified. During study closeout activities, rights should be modified to allow the site only to read data but not enter or change data. Once the required archives have been created, sent, and confirmed received by the site, all access (including view access) to the corresponding data in the EDC system can be removed. Prior to this revocation, the site must continue to have access to the study database in the event of a site inspection or audit. [VI]
15) Evaluation and Continuous Quality Improvement
Understandably in early EDC studies, evaluation questionnaires were frequently performed.31,47,48 Other methods of obtaining feedback include monitoring and analyzing support tickets and meeting minutes and encouraging noting and reporting of problems and suggestions for improvement. EDC is not stagnant and neither is the environment in which it operates. Though there are multiple data sources and data collection methods, web-based EDC will be a mainstay of clinical research data collection for the foreseeable future. As such, organizations should devote attention to improvement of the technology and processes, especially the user experience at the sites.37,49,50
16) Study Archiving – Generating Archive Media for Sites
At the conclusion of a study, the sites must be provided a copy of data they collected throughout the study.5 [I]
A final review of all study documentation should be performed to ensure defined processes have been adhered to such that end of study deliverables from the EDC system meet the requirements specified in applicable regulatory guidance documents. This review should ensure that the study documentation address expectations from regulatory agencies. Some of the guidance and specifications for this review include Guidance for Industry: Providing Regulatory Submissions in Electronic Format – Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications and ICH M2 EWG Electronic Common Technical Document Specification.51,52
a) Quality Review and Replacement of Subject Data
Subject data should present the data collected in a CRF system organized by subject identifier, visit, and form. The data are typically presented in a manner that allows for effective navigation of the subject profile in a format such as a PDF file or a similar format. The subject data should also include an audit trail, electronic signature, and query information, which allows a reviewer the ability to see all data entry and modification that have occurred since it was created. Subject data should be provided on durable media. A master copy of the durable media should be created to contain all the subject profile data. In addition to this master copy, individual copies or other durable media with site-specific data should be created and forwarded accordingly.
b) Archive Media
After the study is complete and a copy of subject data have been generated and securely (e.g., password protected) distributed to sites successfully, a complete copy of the study database should be created for archival purposes.53 [I] The production of archival media is a prerequisite of EDC decommission.
17) Recommended SOPs
a) Recommended Central Standard Operating Procedures
Data Management Plan
User Training
User Management and Security
Study/CRF and Edit Check Change Control
Software System Change Control
Maintenance of Coding Dictionaries
Data Acquisition and Processing
Study Closeout
Database Lock
Generation and Review of Archive Media
b) Recommended SOPs to be in place at Clinical Sites:54
System Setup/Installation
Data Collection and Handling
System Maintenance
Data Backup, Recovery, and Contingency Plans
Security
Change Control
Rusnak provides a good outline for a site SOP covering these topics.54
18) Literature Review
This revision is based on a systematic review of the peer-reviewed literature indexed for retrieval. The goals of this literature review were to (1) identify published research results and reports of EDC methods and evaluation and (2) identify, evaluate, and summarize evidence capable of informing the practice of management and closeout of studies using web-based EDC.
The following PubMed query was used:
(“electronic data capture” OR “EDC” OR (internet AND “data collection”)) AND (“clinical trial” OR “clinical trials” OR “clinical study” OR registry OR registries OR “observational study” OR “interventional study” OR “phase 1” OR “phase 2” OR “phase 3” OR “phase 4” OR “phase I” OR “phase II” OR “phase III” OR “phase IV” OR “first in man” OR “clinical research” OR “device study” OR “interventional trial” OR “phase 1” OR “phase 2” OR “phase 3” OR “phase 4” OR “phase I” OR “phase II” OR “phase III” OR “phase IV” OR RCT OR “randomized clinical trial” OR “non-interventional” OR “post-marketing authorization” OR “post authorization” OR “adaptive trials” OR “feasibility study” OR “phase 2/3” OR “phase II/III” OR “phase 2a” OR “phase 2b” OR “phase IIa” OR “phase IIb” OR “phase IIb/IIIa” OR “phase 2b/3a”)
The search query was customized for and executed on the following databases: PubMed (777 results); CINAHL (230 results); EMBASE (257 results); Science Citation Index/Web of Science (393 results); Association for Computing Machinery (ACM) Guide to the Computing Literature (115 results). A total of 1772 works were identified through the searches. The latest search was conducted on February 8, 2017. Search results were consolidated to obtain a list of 1368 distinct articles. Because this was the first review for this chapter, the searches were not restricted to any time range. Literature review and screening details are included in the PRISMA diagram for the chapter.
Two reviewers used inclusion criteria to screen all abstracts. Disagreements were adjudicated by the writing groups. Forty-nine meeting inclusion criteria were selected for review. The selected articles were read by the writing group and 104 additional sources identified through the review. Each of the 153 articles was read for mention of explicit practice recommendations or research results informing practice. A total of 82 articles were identified as relevant to the history of EDC or to one or more of the EDC GCDMP Chapters. Fourteen articles provided evidence for this EDC Chapter. Relevant findings from these 14 articles have been included in the chapter and graded according to the GCDMP evidence grading criteria in the table below. This synthesis of the literature relevant to web-based EDC was performed to support the transition of the EDC chapters to an evidence-based guideline.
Articles supporting assertions or otherwise informing practice recommendations in GCDMP chapters are graded according to the strength of the evidence. The GCDMP has adopted the following grading criteria.
Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) For Web-based EDC Study Conduct, Maintenance, and Closeout.
19) Revision History
| Date | Revision description |
|---|---|
| September 2003 | Initial publication as Electronic Data Capture Principles. |
| May 2007 | Revised for style, grammar, and clarity. Substance of chapter content unchanged. |
| September 2008 | Revised to reflect the orientation of chapter towards the conduct phase of EDC. Content updated and organization of material revised. Study concept and start up, and study closeout content moved to separate chapters. |
| May 2020 | Content updated and organization of material revised. Study conduct was combined with study closeout into one comprehensive chapter. |
Competing Interests
The authors have no competing interests to declare.
References
1. Marks, RG. Validating electronic source data in clinical trials. Control Clin Trials. 2004; 25: 437–446. DOI: http://doi.org/10.1016/j.cct.2004.07.001
2. Food and Drug Administration. US Department of Health and Human Services. ICH E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1), March 2018. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r2-good-clinical-practice-integrated-addendum-ich-e6r1.
3. Food and Drug Administration. US Department of Health and Human Services. Electronic Records; Electronic Signatures, 21 CFR §11 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
4. Food and Drug Administration. US Department of Health and Human Services. Guidance for Industry: Computerized Systems Used in Clinical Investigations. May, 2007. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computerized-systems-used-clinical-investigations.
5. Food and Drug Administration. US Department of Health and Human Services. Investigational New Drug Application; Investigator recordkeeping and record retention, 21 CFR §312.62 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
6. Food and Drug Administration. US Department of Health and Human Services. New animal drugs for investigational use; New animal drugs for investigational use exempt from section 512(a) of the Federal Food, Drug, and Cosmetic Act. 21 CFR §511.1(b) (7) (ii) ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
7. Food and Drug Administration. Department of Health and Human Services, Investigational device exemptions; Records. 21 CFR §812.140 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
8. Medicines & Healthcare products Regulatory Agency (MHRA). ‘GXP’ Data Integrity Guidance and Definitions. Revision 1: March 2018. Available at https://www.gov.uk/government/publications/guidance-on-gxp-data-integrity.
9. Food and Drug Administration. US Department of Health and Human Services, General Principles of Software Validation; Final Guidance for Industry and FDA Staff, January 2002, available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation.
10. European Commission Health and Consumers Directorate-general. EudraLex; The Rules Governing Medicinal Products in the European Union Volume 4: Annex 11: Computerised Systems ( 2011). Available at https://ec.europa.eu/health/documents/eudralex/vol-4_ga.
11. GAMP® 5 A Risk-based Approach to Compliant GxP Computerized Systems. North Bethesda, MD: International Society for Pharmaceutical Engineering (ISPE). 2008.
12. Food and Drug Administration. US Department of Health and Human Services. Guidance for industry: Use of Electronic Health Record Data in Clinical Investigations. July 2018. Accessed September 17, 2020. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-electronic-health-record-data-clinical-investigations-guidance-industry.
13. Food and Drug Administration. US Department of Health and Human Services. Guidance for Industry: Electronic Source Data in Clinical Investigations. September 2013. Accessed August 8, 2018. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electronic-source-data-clinical-investigations.
14. Unützer J, Choi Y, Cook IA, Oishi S. A web-based data management system to improve care for depression in a multicenter clinical trial. Psychiatr Serv. 2002 Jun; 53(6): 671–673. 678. DOI: http://doi.org/10.1176/ps.53.6.671
15. Mitchel J, Ernst C, Cappi S, et al. Implementing internet-based clinical trials. DIA Forum. 2004; 40: 22–23.
16. Mitchel JT, Kim YJ, Choi J, et al. The impact of electronic data capture on clinical data management perspectives from the present into the future. MONITOR. 2008 August.
17. Summa W. Electronic data capture: Automated management of clinical trial data. Pharmind. 2004; Special Issue 5a/.
18. Litchfield J, Freeman J, Schou H, Elsley M, Fuller R, Chubb B. Is the future for clinical trials internet-based? A cluster randomized clinical trial. Clin Trials. 2005; 2: 72–79. DOI: http://doi.org/10.1191/1740774505cn069oa
19. Wilkinson M, Young R, Harper B, Machion B, Getz K. Baseline assessment of the evolving 2017 eClinical Landscape. Therapeutic Innovation & Regulatory Science. 2018; 1–10. DOI: http://doi.org/10.1177/2168479018769292
20. Mitchel JT, Kim YJ, Choi J, Hays V, Langendorf J, Cappi S. Impact of IBCTs on clinical trial efficiency. ACT. 2006 August.
21. Richardson A. Planning and running the eClinical trial. ACT. 2003 January.
22. Cramon P, Rasmussen AK, Bonnema SJ, et al. Development and implementation of PROgmatic: A clinical trial management system for pragmatic multi-centre trials, optimised for electronic data capture and patient-reported outcomes. Clin Trials. 2014; 11: 344–354. DOI: http://doi.org/10.1177/1740774513517778
23. Mitchel J, Weingard K, Schloss Markowitz J, et al. How direct data entry at the time of the patient visit is transforming clinical research – perspective from the clinical trial research site. INSITE. 2013; 2nd Quarter: 40–43.
24. Mitchel JT, Cho T, Gittleman DA, et al. Three pronged approach to clinical trial monitoring. ACT. 2014 June/July.
25. Zozus, M. The Data Book: Collection And Management Of Research Data. 1st ed. Boca Raton, FL: Taylor & Francis/CRC Press; 2017. DOI: http://doi.org/10.1201/9781315151694
26. Mitchel J, You J, Lau A, et al. Clinical Trial Data Integrity. Using internet-based remote data entry to collect reliable data. Appl Clin Trials. 2003 March: (Supplement, 6–8).
27. Mitchel J, You J, Kim YJ, et al. Internet-based clinical trials—practical considerations. Pharmaceutical Development and Regulations. 2003; 1: 29–39. DOI: http://doi.org/10.1007/BF03257363
28. Marks RG, Conlon M, Ruberg SJ. Paradigm shifts in clinical trials enabled by information technology. Stat Med. 2001; 20: 2683. DOI: http://doi.org/10.1002/sim.736
29. Marks R, Bristol H, Conlon M, Pepine CJ. Enhancing clinical trials on the internet: lessons from INVEST. Clin Cardiol. 2001; 24(11 Suppl): V17–V23. DOI: http://doi.org/10.1002/clc.4960241707
30. Pellegrino T. Supporting EDC in a clinical trial environment. Appl Clin Trials. 2002; (Aug. 2) (SDC Suppl.): 16–9.
31. Dimenas E, Johansson D, Palmblad M. Wrangstadh M. Clinical Operations Online (COOL)-a world wide web-based approach to running clinical trials: Results from two international multicenter gastrointestinal trials. Drug Inf J. 2001; 35: 745–753. DOI: http://doi.org/10.1177/009286150103500313
32. Mitchel JT, Markowitz JMS, Yin H, et al. Lessons learned from a direct data entry phase 2 clinical trial under a US investigational new drug application. Drug Inf J. 2012; 46(4): 464–471. DOI: http://doi.org/10.1177/0092861512449530
33. Mitchel JT, Dean Gittleman D, Park G, et al. The impact on clinical research sites when direct data entry occurs at the time of the office visit: A tale of 6 studies. InSite. 2014; Second Quarter.
34. Arndt S, Tyrell G, Woolson RF, Flaum M, Andreasen NC. Effects of errors in a multicenter medical study: preventing misinterpreted data. J Psych Res. 1994; 28: 447–459. DOI: http://doi.org/10.1016/0022-3956(94)90003-5
35. Abouelenein S, Williams TB, Baldner J, Zozus MN. Analysis of professional competencies for the clinical research data management profession. Data Basics. 2020; 26(1): 6–21. Available at www.scdm.org.
36. Food and Drug Administration. US Department of Health and Human Services. Guidance for industry: Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring. August 2013. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/oversight-clinical-investigations-risk-based-approach-monitoring.
37. Helms RW. Data quality issues in electronic data capture. Drug Inf J. 2001; 35: 827–837. DOI: http://doi.org/10.1177/009286150103500320
38. Nahm ML, Pieper CF, Cunningham MM. Quantifying data quality for clinical trials using electronic data capture. PLoS One. 2008; 3(8): e3049. DOI: http://doi.org/10.1371/journal.pone.0003049
39. US Department of Health and Human Services. Health Insurance Portability and Accountability Act of 1996, (HIPAA) Public Law 104-191, as amended, 42 United States Code 1320-d. 1996. Available at: https://www.govinfo.gov/content/pkg/PLAW-104publ191/html/PLAW-104publ191.htm
40. Official Journal of the European Communities. Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995, Article 8. No. L 281/31. 1995. Available at https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A31995L0046
41. Food and Drug Administration. US Department of Health and Human Services. Applications for FDA Approval to Market a New Drug, 21 CFR §314 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm
42. Mitchel J, You J, Lau A, Kim YJ. Paper versus web; A tale of three trials. Appl Clin Trials. 2001; August: 34–35.
43. Spink C. Electronic Data Capture (EDC) as a means for e-clinical trial success, IBM Global Services (report BIWUK00002-2-0202-AP). Pharmaceutical Clinical Development. 2002; March.
44. Meadows B. A comparison of paper-based data submission to remote data capture for minimizing data entry errors in cancer clinical research. 2006. University of Maryland Baltimore.
45. Zozus NM, Kahn M, Weiskopf N. Clinical research data quality. Clinical Research Informatics, Health Informatics. Richesson RL, Andrews JE (eds.), 2018; 175–201. 2nd ed. London: Springer-Verlag.
46. Zozus MN, Pieper C, Johnson CM, et al. Factors affecting accuracy of data abstracted from medical records. PLoS ONE. 2015; 10(10): e0138649. PMC4615628. DOI: http://doi.org/10.1371/journal.pone.0138649
47. Lopez-Carrero C, Arriaza E, Bolanos E, et al. Internet in clinical research based on a pilot experience. Contemp Clin Trials. 2005; 26(2): 234–43. DOI: http://doi.org/10.1016/j.cct.2004.11.017
48. Pavlović I, Miklavcic D. Web-based electronic data collection system to support electrochemotherapy clinical trial. IEEE Trans Inf Technol Biomed. 2007 Mar; 11(2): 222–230. DOI: http://doi.org/10.1109/TITB.2006.879581
49. Bunn G. Scaling up EDC. How to move away from paper trials. Appl Clin Trials. 2002; (Aug. 2); (SDC Supl): 12–4.
50. MacGarvey A. EDC. State of the Art. Innovations in Pharmaceutical Technology. 2005; pp. 116–118.
51. Food and Drug Administration. US Department of Health and Human Services. Guidance for Industry: Providing Regulatory Submissions in Electronic Format – Certain Human Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications. February 2020. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/providing-regulatory-submissions-electronic-format-certain-human-pharmaceutical-product-applications.
52. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH M2 EWG Electronic Standards for the Transfer of Regulatory Information – ESTRI. Electronic Common Technical Document Specification, V 3.2.2, July 2008. Available at https://www.ich.org/page/ich-electronic-common-technical-document-ectd-v322-specification-and-related-files.
53. Food and Drug Administration. US Department of Health and Human Services, Investigational New drug Application; Recordkeeping and record retention. 21 CFR §312.57 ( 1997). Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm.
54. Rusnak E. SOPs for electronic records and data collection in clinical research. Issues in clinical trials management. Research Practitioner. 2001; 2(5).