
1) Learning Objectives
After reading this chapter, the reader should understand
the importance of validating computer systems that are used in the conduct of clinical studies
the stages of Software Development Life Cycle (SDLC), from planning through decommissioning
the validation activities associated with software that is developed in-house, vendor-licensed, and open-source
the validation activities associated with study-specific configurations of applications
a risk-based approach to software development and validation
the necessary documentation that is part of the SDLC package and the archival plan for those deliverables
change control requirements needed to maintain the validated state of the system
the use of real-world data and associated validation requirements
recommended standard operating procedures (SOPs) for SDLC.
2) Introduction
Clinical research has transitioned in the last 25+ years from paper-based processes to electronic data collection tools. Since 21 CFR Part 11 became effective in 1997, determination is made when the US Food and Drug Administration (FDA) will accept an electronic record or signature instead of a handwritten record or signature. “To comply with Part 11, computer systems that create, modify, maintain, or transmit electronic records subject to FDA predicate rules must be validated (21 CRF 11.10[a]).” [VII] (Advarra 2020)1
Software should provide valuable solutions to users. In clinical research, the integrity of the data is at the very core of its value. To have confidence in the clinical data, there must be confidence in the system and structure in which the data are collected and processed. To that end, developing the software used to produce that data necessitates identifying requirements and detailed planning of its life cycle.
SDLC in clinical research provides structure, control, and assurance that the software complies with the technical and regulatory requirements and meets the need for its intended purpose. [VII] (Advarra 2020)1
User experience should be taken into account when developing the software, so that data are recorded without difficulty and with accuracy. System testing minimizes risks, ensures functionality, and provides the necessary verification that the data are reliable. Change control procedures help to maintain the validated state of the system. Per FDA guidance, the ALCOA principles of data integrity (attributable, legible, contemporaneous, original, accurate) should be applied; an established SDLC will meet these deliverables to produce quality clinical data.2
3) Scope
This chapter addresses SDLC activities with a risk-based approach that should accompany the planning, installation, validation, maintenance, and decommissioning of computerized systems that are used in the conduct of a clinical study (eg, electronic data capture systems and applications in which clinical study data are collected, stored, and managed), as well as the validation tasks involved when designing study-specific applications. Included in the chapter will be software that is developed in-house, licensed by a vendor, and open-source. Although data collection tools encompass a large variety of sources (eg, surveys, devices, ePRO, to name just a few), the general principles can be applied to ensure the software and applications meet their requirements and specifications and are suitable for intended use.
Software/firmware developed as part of a medical device is not covered in this chapter.
Although some of the specific topics addressed by this chapter may not be the direct responsibility of Clinical Data Management (CDM) personnel, CDM must have an ongoing awareness of the requirements and ensure tasks have been completed in accordance with the principles and standards of their organization, regulatory bodies, and good clinical practice.
4) Minimum Standards
Validation of system(s) used as part of clinical studies is an integral part in ensuring quality, integrity, and interpretability of the data. The International Council for Harmonisation (ICH) E6 (R2)3 contains several passages particularly relevant to electronic data capture (EDC) software development and validation.
Section 1.65 describes Validation of Computerized Systems as “a process of establishing and documenting that the specified requirements of a computerized system can be consistently fulfilled…. from design until decommissioning of the system or transition to a new system.”3
Section 2.8 states, “Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).”3
Section 2.10 states, “All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification.”3
Section 2.13 indicates that system procedures ensuring trial quality should be implemented to secure protection of human subjects and data quality.3
Section 5.0 states, “The methods used to assure and control the quality of the trial should be proportionate to the risks inherent in the trial and the importance of the information collected.”3
Section 5.5.3 relates to validation of computerized systems and states, “When using electronic trial data handling and/or remote electronic trial data systems, the sponsor should, a) Ensure and document that the electronic data processing system(s) conforms to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (i.e., validation).”3 This requirement echoes Title 21 CFR Part 11 and requires that the installation of the EDC system used for a study be validated.
Section 5.5.3’s first addendum states that validation of computer systems should be risk-based. “The sponsor should base their approach to validation of such systems on a risk assessment that takes into consideration the intended use of the system and the potential of the system to affect human subject protection and reliability of trial results.”3 This general Good Clinical Practice (GCP) requirement promotes right-sizing the type and extent of validation of system functionality to the assessed risk associated with functionality. In EDC systems, building a study within validated software has significantly less risk than developing new software. Open-source software has different risks than commercial software or in-house custom-developed software. These risk differences are considerations in EDC software selection and initial implementation including system validation.
Section 5.5.3 addendum b states that an organization “Maintains SOPs for using these systems.”3 The 5.5.3 addendum c-h introductory statement enumerates topics that should be covered in SOPs. “The SOPs should cover system setup, installation, and use. The SOPs should describe system validation and functionality testing, data collection and handling, system maintenance, system security measures, change control, data backup, recovery, contingency planning, and decommissioning.”3 These requirements apply to system selection and initial implementation in that the processes covered by the requirement can be significantly impacted by the functionality available in an EDC system being used by a sponsor. Further, individual requirements are addressed in the section, such as 5.5.3 addendum (e) “Maintain a list of the individuals who are authorized to make data changes”, (g) “Safeguard the blinding, if any,” and (h) “Ensure the integrity of the data, including any data that describe the context, content, and structure.”3
Data can be collected using the electronic health records (EHR) system as part of clinical trials. One can solely collect via EHR or integrated with EDC data collection. The FDA guidance on Use of Electronic Health Record Data in Clinical Investigations4 contains several references relevant in the validation of data used in EHR:
Section IV: “Interoperable systems allow electronic transmission of relevant EHR data to the EDC system… Interoperable systems may also reduce errors in data transcription, allowing for the improvement in data accuracy and the quality and efficiency of the data collected in clinical investigations.”4
Section IV.A: “The data exchange between EHR and EDC systems should leverage the use of existing open data standards, when possible, while ensuring that the integrity and security of data are not compromised.”4
Section IV.C: “Sponsors should ensure that the interoperability of EHR and EDC systems (e.g., involving the automated electronic transmission of relevant EHR data to the EDC system) functions in the manner intended in a consistent and repeatable fashion and that the data are transmitted accurately, consistently, and completely.”4
Section IV.C: “FDA encourages sponsors to periodically check a subset of the extracted data for accuracy, consistency, and completeness with the EHR source data and make appropriate changes to the interoperable system when problems with the automated data transfer are identified.”4
Section V.C: “FDA does not intend to assess EHR systems for compliance with 21 CFR part 11. However, part 11 applies to the sponsor’s EDC system that extracts the EHR data for use in a clinical investigation, and FDA intends to assess the sponsor’s EDC system for compliance with part 11, as provided in the guidance for industry Part 11, Electronic Records; Electronic Signatures – Scope and Application.”4
Title 21 CFR Part 11 also identifies regulatory requirements for traceability, training, and qualification of personnel, and for the validation of computer systems used in clinical studies.
Personnel should meet training requirements (21 CFR Part 11).2
Sec. 11.10 Controls for closed systems: “Persons who use closed systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, when appropriate, the confidentiality of electronic records, and to ensure that the signer cannot readily repudiate the signed record as not genuine. Such procedures and controls shall include the following:
(e) Use of secure, computer-generated, time-stamped audit trails to independently record the date and time of operator entries and actions that create, modify, or delete electronic records. Record changes shall not obscure previously recorded information. Such audit trail documentation shall be retained for a period at least as long as that required for the subject electronic records and shall be available for agency review and copying.
(g) Use of authority checks to ensure that only authorized individuals can use the system, electronically sign a record, access the operation or computer system input or output device, alter a record, or perform the operation at hand.
(h) Use of device (e.g., terminal) checks to determine, as appropriate, the validity of the source of data input or operational instruction.”2
Sec. 11.30 Controls for open systems: “Persons who use open systems to create, modify, maintain, or transmit electronic records shall employ procedures and controls designed to ensure the authenticity, integrity, and, as appropriate, the confidentiality of electronic records from the point of their creation to the point of their receipt.”2
Sec. 11.50 Signature manifestations: “(a) Signed electronic records shall contain information associated with the signing that clearly indicates all of the following: (1) The printed name of the signer; (2) The date and time when the signature was executed; and (3) The meaning (such as review, approval, responsibility, or authorship) associated with the signature.”2
EU GMP Annex 11 Section on data integrity states: “Data integrity, reliability and robustness will depend on the design and the validation status of the computerised systems used. Failure to document and therefore demonstrate the validated state of a computerised system is likely to pose a risk to data integrity, reliability and robustness, which depending on the criticality of the affected data may result in a recommendation from the GCP inspectors to the CHMP not to use the data within the context of an MAA.”5 For reference, MAA refers to Marketing Authorization Application.
“It is not acceptable to use computerised systems in clinical trials for which the validation status is not confirmed or for which appropriate documentation on system validation cannot be made available to GCP inspectors.”5
The GAMP 5 A Risk-based Approach to Compliant GxP Computerized Systems6 document provides guidance on the activities required during the SDLC to help establish the GxP computerized system(s) is fit for use as intended. This document covers the different phases of the life cycle starting at the planning phase with the functional/user requirements, design, testing/verification, deployment and maintenance phases. It also refers to the roles and responsibilities involved in planning, verification and deployment of computerized systems and covers how in-house system(s), vendor selected product(s), open-source system(s) are to be appraised in the verification process, while evaluating it in a risk-based approach. The GAMP 5 contains the following relevant references to ensure systems are fit for intended use:
Section 2.1.3: “Life cycle activities should be scaled according to:
System impact on patient safety, product quality and data integrity (Risk Assessment)
System complexity and novelty (architecture and categorization of system components)
Outcome of supplier assessment (supplier capability)”6
Section 2.1.4: “Qualitative or Quantitative techniques may be used to identify and manage risks.”6
Section 2.1.5: Leveraging Supplier Involvement. “Planning should determine how best to use supplier documentation, including existing test documentation, to avoid wasted effort and duplication. Justification for the use of supplier documentation should be provided by the satisfactory outcome of supplier assessments, which may include supplier audits.”6
Section 3.1: “An inventory of computerized systems should be maintained.”6
Section 4.2.4: “At the conclusion of the project, a computerized system validation report should be produced summarizing the activities performed, any deviations from the plan, any outstanding and corrective actions, and providing a statement of fitness for intended use of the system.”6
Section 4.2.4: “Well managed system handover from the project team to the process owner, system owner, and operational user is a pre-requisite for effectively maintaining compliance of the system during operation.”6
Section 4.2.5.2: “Change management procedures also should be established.”6
Section 4.2.5.2: “Any involvement of the supplier in these processes should be defined and agreed.”6
Section 4.3.4.1: “Change management is a critical activity that is fundamental to maintaining the compliant status of systems and processes… The process should ensure that changes are suitably evaluated, authorized, documented, tested and approved before implementation and subsequently closed.”6
Section 4.3.5: “Electronic data archives holding GxP data from retired systems also should be subject to periodic review.”6
Section 4.3.6.1: “Processes and procedures should be established to ensure that backup copies of software, records, and data are made, maintained, and retained for a defined period within safe and secure areas.”6
Section 4.3.6.2: “A Business Continuity Plan defines how the business may continue to function and handle data following failure. (i.e.: Disaster recovery planning)”6
Section 4.3.7.1: “Role-based security should be implemented, if possible, to ensure that sensitive data and functions are not compromised.”6
Section 6.1.1: “Each regulated company should have a defined policy for ensuring that computerized systems are compliant and fit for intended use.”6
Even though the General Principle of Software Validation – Final Guidance for Industry and FDA Staff7 is targeted for software used in medical devices, some of the concepts defined in this guidance can be applied to other software used in clinical studies. The following sections are important in any software development life cycle:
Section 3.5: “The software validation process cannot be completed without an established software requirements specification (Ref: 21 CFR 820.3(z) and (aa) and 820.30(f) and (g)).”7
Section 4.7: “Whenever software is changed, a validation analysis should be conducted not just for validation of the individual change, but also to determine the extent and impact of that change on the entire software system.”7
Section 4.9: “When possible, an independent evaluation is always better, especially for higher risk applications.”7
Section 4.9: “Another approach is to assign internal staff members that are not involved in a particular design or its implementation, but who have sufficient knowledge to evaluate the project and conduct the verification and validation activities.”7
Section 5.1: “Management must identify and provide the appropriate software development environment and resources.”7
Section 5.2.5: “An essential element of a software test case is the expected result. It is the key detail that permits objective evaluation of the actual test result.”7
Section 5.2.5: “Testing of a changed software product requires additional effort. Not only should it demonstrate that the change was implemented correctly, but testing should also demonstrate that the change did not adversely impact other parts of the software product.”7
Section 5.2.5: “Test procedures, test data, and test results should be documented in a manner permitting objective pass/fail decisions to be reached. They should also be suitable for review and objective decision making subsequent to running the test, and they should be suitable for use in any subsequent regression testing.”7
Section 6: “When computers or automated data processing systems are used as part of production or the quality system, the [device] manufacturer shall validate computer software for its intended use according to an established protocol. (See 21 CFR §820.70(i)).”7
Section 6: “Software tools are frequently used to design, build, and test the software that goes into an automated medical device…. All of these applications are subject to the requirement for software validation, but the validation approach used for each application can vary widely.”7
The Guidance for Industry – Computerized Systems Used in Clinical Investigations8 outlines how computerized systems should be used in clinical trials to maintain data integrity of electronic data from its creation, modification, maintenance, and retirement until it is used for submission. Electronic data is defined as a record transcribed from hard copy source document to an electronic system, a direct entry or automatically recorded in a computerized system. Medical devices not covered in this chapter are also not covered in this guidance.
Section IV.A: “The computerized systems should be designed: (1) to satisfy the processes assigned to these systems for use in the specific study protocol (e.g., record data in metric units, blind the study), and (2) to prevent errors in data creation, modification, maintenance, archiving, retrieval, or transmission.”8
Section IV. B: “There should be specific procedures and controls in place when using computerized systems to create, modify, maintain, or transmit electronic records, including when collecting source data at clinical trial sites.”8
Section IV.C: “When original observations are entered directly into a computerized system, the electronic record is the source document. Under 21 CFR 312.62, 511.1(b)(7)(ii) and 812.140, the clinical investigator must retain records required to be maintained under part 312, § 511.1(b), and part 812, for a period of time specified in these regulations.”8
Section IV D.1: “Access must be limited to authorized individuals (21 CFR 11.10(d).”8
Section IV D.3: “Controls should be established to ensure that the system’s date and time are correct.”8
Section IV E: “In addition to internal safeguards built into a computerized system, external safeguards should be put in place to ensure that access to the computerized system and to the data is restricted to authorized personnel.”8
Section IV F.2: “The computerized system should be designed in such a way that retrieved data regarding each individual subject in a study is attributable to that subject.”8
Section IV F.3: “For each study, documentation should identify what software and hardware will be used to create, modify, maintain, archive, retrieve, or transmit clinical data.”8
Section IV F.4: “Sufficient backup and recovery procedures should be designed to protect against data loss. Records should regularly be backed up in a procedure that would prevent a catastrophic loss and ensure the quality and integrity of the data. Records should be stored at a secure location.”8
Section IV F.5: “The effects of any changes to the system should be evaluated and some should be validated depending on risk.”8
Section IV G: “Training should be provided to individuals in the specific operations with regard to computerized systems that they are to perform. Training should be conducted by qualified individuals on a continuing basis, as needed, to ensure familiarity with the computerized system and with any changes to the system during the course of the study.”8
Guidance for Industry: Electronic Source Data in Clinical Investigations9 Electronic source data comes in many forms, and we must understand the type of record in question, and the requirements to safeguard the authenticity and integrity of that record.
In the background section of the guidance it states: “electronic record as any combination of text, graphics, data, audio, pictorial, or other information represented in digital form that is created, modified, maintained, archived, retrieved, or distributed by a computer system.”9
It also states that “Source data should be attributable, legible, contemporaneous, original, and accurate (ALCOA) and must meet the regulatory requirements for recordkeeping.”9 This includes all data that may be collected as source during the course of a clinical study. Source data is also defined in this guidance as “all information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical investigation used for reconstructing and evaluating the investigation.”9 Thus, all data initially recorded in an electronic system is defined as electronic source data (eg, EHR, tablet electronic case report forms (eCRF), diaries, questionnaires).
In section III.A.2, the guidance refers to different ways to collect the data directly and it may include requirements that roles are applied appropriately based on what electronic source data is entered in a specific CRF. It is critical for a CDM to understand exactly where source data is coming from as specific rights access may need to be validated at a study specific setup.
In section III.A.2.a, the guidance indicates, “This direct entry of data can eliminate errors by not using a paper transcription step before entry into the eCRF. For these data elements, the eCRF is the source. If a paper transcription step is used, then the paper documentation should be retained and made available for FDA inspection (see section III.A.2.c).”9
As a CDM, it is important to understand the originator of the source data to consider the validation steps necessary, in section III A.1 specifically around security of the data.
“A list of all authorized data originators (i.e., persons, systems, devices, and instruments) should be developed and maintained by the sponsor and made available at each clinical site.”9
“Controls must be employed to ensure the security and integrity of the authorized user names and password. When electronic thumbprints or other biometric identifiers are used in place of an electronic log-on/password, controls should be designed to ensure that they cannot be used by anyone other than their original owner”9 to be in compliance with 21 CFR Part 11. It is important to understand what security system is in place and what validation requirements have already been performed by the vendor or what may be required at study start up.
“When a system, device, or instrument automatically populates a data element field in the eCRF, a data element identifier (see section III.A.3) should be created that automatically identifies the particular system, device, or instrument (e.g., name and type) as the originator of the data element.”9
Section III A.2.d.: “Unlike a direct transmission to an eCRF from instruments or medical devices, EHRs can use intervening processes (e.g., algorithms for the selection of the appropriate data elements).”9 EHR systems are not required to be validated as described in 21 CFR Part 11, but the interoperability with an EDC system shall meet the 21 CFR Part 11.9 The interoperability requirements are discussed later within the EHR section of this chapter.
Section III A.3 and Section III A.4 describe the audit trail requirements (originator, date, and timestamp of changes) of electronic source data. These requirements may be validated by the vendor and/or sponsor depending on the system and/or its integration. As a CDM, understanding the data flow of these systems is important to address potential study specific validation requirements. “These data element identifiers will allow sponsors, FDA, and other authorized parties to examine the audit trail of the eCRF data (and this audit trail should be readily available in a human readable form).”9 “Only a clinical investigator(s) or delegated clinical study staff should perform modifications or corrections to eCRF data. Modified and/or corrected data elements must have data element identifiers that reflect the date, time, originator and reason for the change, and must not obscure previous entries. A field should be provided allowing originators to describe the reason for the change (e.g., transcription error). Automatic transmissions should have traceability and controls via the audit trail to reflect the reason for the change.”9 These items should be part of user requirements for the audit trail validation of the system or the interoperability of systems. The guidance even states that “the eCRF system should include a functionality that enables FDA to reveal or access the identifiers related to each data element.”9
Section III.A.5 provides additional information on the recommendation when using a system for electronic source. “We encourage the use of electronic prompts, flags, and data quality checks in the eCRF to minimize errors and omissions during data entry”.9 These prompts are made available to the data originator to avoid errors and to give the opportunity to correct these errors at the time of entry of the electronic source.
Section III B.1.a also refers to the investigator’s requirement for acknowledging review of the data using electronic signature. “To comply with the requirement to maintain accurate case histories clinical investigator(s) should review and electronically sign the completed eCRF for each subject before the data are archived or submitted to FDA. Use of electronic signatures must comply with part 11 (21 CFR Part 11).”2, 9
Section III D indicates “The sponsor should have a list (e.g., in a data management plan) of the individuals with authorized access to the eCRF.”9 and also corroborates ICH E63 Section 2.8 on training and Title 21 CFR Part 11 on access control. “Only those individuals who have documented training and authorization should have access to the eCRF data. Individuals with authorized access should be assigned their own identification (log-on) codes and passwords. Log-on access should be disabled if the individual discontinues involvement during the study.”9
Section IV also supports the information provided in Use of Electronic Health Record Data in Clinical Investigations where the “FDA does not intend to assess the compliance of EHRs with part 11.”4, 9
With these requirements in mind, in Table 1 we state the following minimum standards for the software development and validation of computerized systems utilized in clinical studies.
Minimum Standards.
|
5) Best Practices
Design a Validation Plan that defines the strategy for the software validation. [VI]
Use a risk management approach for SDLC, taking into account patient safety, data integrity and product quality. (2011 Von Culin) [VII]10
Define the requirements necessary for the software to perform the way it is expected. (GAMP 5) [III]6
Devise a Test Plan to ensure the software conforms with the stated requirements. (GAMP 5) [III]6
Ensure the system has a Traceability Matrix linking test cases to requirements. [VI]
Ensure automated testing tools and test environments have documented assessments of their adequacy. (2016 Nidagundi) [III]11
If data are transferred to another data format or system, include scripts that demonstrate data are not altered in value and/or meaning during this transfer process. [VI]
Summarize testing activities and results in a Testing Summary Report. [VI]
Write a Software Verification and Validation Summary Report with documented approval for the release of the software into production. (GAMP Good Practice Guide Testing of GxP systems, Appendix T7) [III]6
Ensure a security system is in place to protect against unauthorized access, and maintain a list of the individuals authorized to create, access, modify, or delete data. (45 CFR Part 164) [III]12
Adhere to and document a Change Control process to maintain the validated state of the system. [VI]
Conduct a periodic review of the system. (GAMP 5 – section 4.3.5) [III]6
Archive documentation of all stages of the SDLC. [VI]
Provide documented training to ensure users are qualified to perform system tasks. [VI]
Establish a backup/recovery plan for the system. (GAMP 5 Appendix O9 section 4.4) [III]6
Develop a Business Continuity Plan that describes how business would resume following a disruption. (GAMP 5 – Appendix O10 -section 1) [III]6
For vendor-licensed software, evaluate validation documentation/activities to determine qualification and level of additional testing that may be needed by the sponsor. [VI]
For in-house systems, complete all stages of SDLC, including creating training materials and providing technical support. [VI]
For open-source software, develop the software in accordance with an organization’s quality management system and follow a risk-based approach to decide the extent of testing, validation, and documentation. [VI]
For study-specific customization of software, follow the same best practices as validating the software, while determining the degree of testing using a risk-based approach. [VI]
If including Real World Data in the clinical study, document the source and data flow between systems. (Use of Electronic Health Record Data in Clinical Investigations Guidance for Industry (fda.gov) Section V- A) [III]4
Good Clinical Data Management Practices (GCDMP) Evidence Grading Criteria.
| Evidence Level | Evidence Grading Criteria |
|---|---|
| I | Large, controlled experiments, meta, or pooled analysis of controlled experiments, regulation or regulatory guidance |
| II | Small controlled experiments with unclear results |
| III | Reviews or synthesis of the empirical literature |
| IV | Observational studies with a comparison group |
| V | Observational studies including demonstration projects and case studies with no control |
| VI | Consensus of the writing group including GCDMP Executive Committee and public comment process |
| VII | Opinion papers |
6) Software Development Life Cycle
SDLC refers to the methodology used to plan, create, test, deploy, and maintain a software throughout its lifecycle until decommission. This section describes SDLC components that are typically part of any software type (eg, in-house, vendor- licensed, or open-source). There are different methodologies that can be used and combined during SDLC. Refer to GAMP 56 for more information on each methodology, including more common ones such as Waterfall and Agile. Waterfall methodology is an established approach in SDLC and may be more appropriate for larger projects than Agile methodology, which requires more interaction by the different groups involved in the validation of the project, including the QA group [V] (2016 Nidagundi).11
It is also important to note the different terminology commonly used between software verification and software validation. As defined in the “General Principles of Software Validation; Final Guidance of Industry and FDA Staff”7:
Software verification “provides objective evidence that the design outputs of a particular phase of the software development life cycle meet all of the specified requirements for that phase. Software verification looks for consistency, completeness, and correctness of the software and its supporting documentation, as it is being developed, and provides support for a subsequent conclusion that software is validated.”7
Software validation is the “confirmation by examination and provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled.”7
In other words, verification confirms that the output matches the specifications and validation confirms that the specifications meet the intended use. No matter the terminology that is used, it is important that both steps are performed. These steps together may be referred to as “testing” throughout this chapter.
SDLC consists of three stages that are ideally conducted in the following order and pictured in Figure 1:
Installation Qualification (IQ): Evidence that the system installed successfully as per specifications.
Operational Qualification (OQ): Evidence that the system operates as expected as per functional requirements and business processes consistently and accurately in the selected environment.
Performance Qualification (PQ): Evidence that the system operates as expected in the user environment. This is performed by the end user and considered as User Acceptance Testing (UAT).
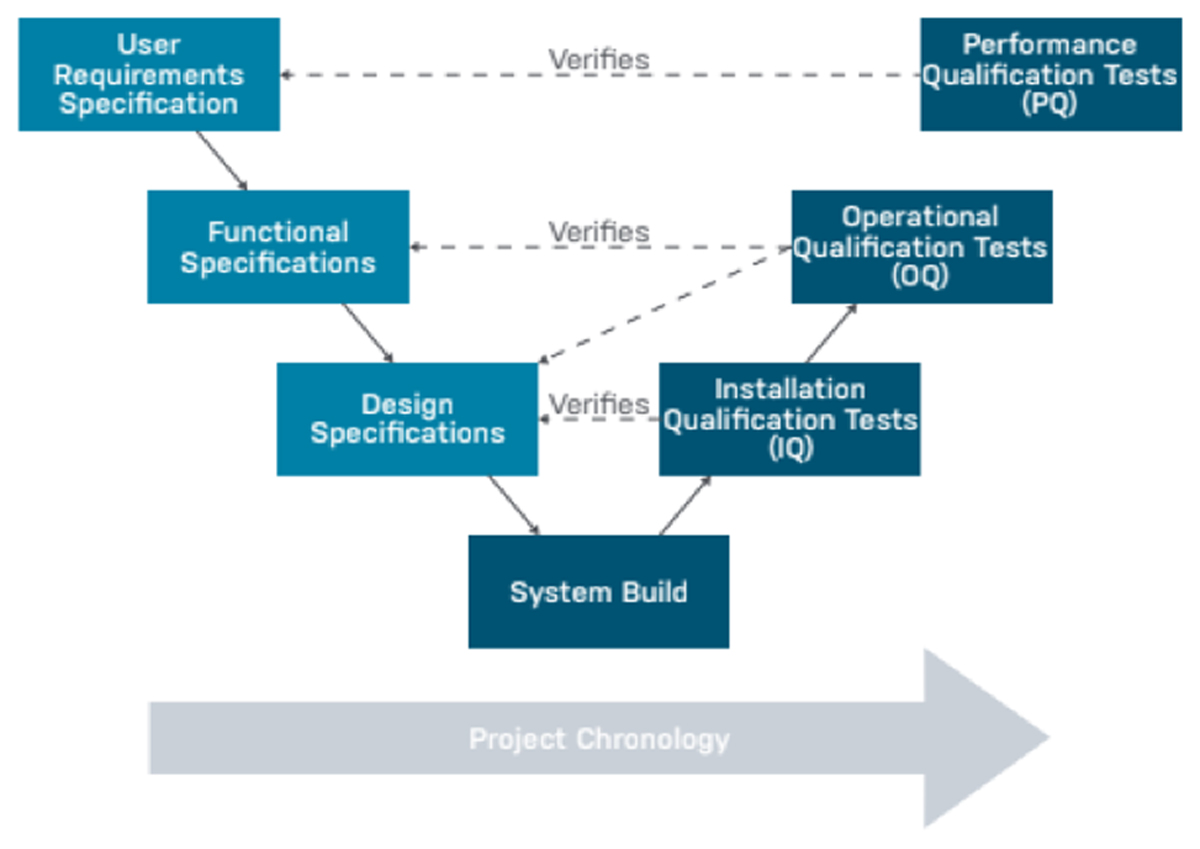
A basic framework from Advarra (2020).1
A CRO/sponsor needs to decide how the software will be used, which will determine the extent of testing required. There is a minimum amount of documentation required to demonstrate that the software functions as intended. This documentation is important to ensure system security and privacy regulations are maintained as well as processing data to maintain its accuracy and integrity from source.
Computer system validation deliverables should be archived (stored for ready retrieval in a secure, limited access manner) for as long as the necessary retention limits for the records associated with the system apply, as required by applicable regulations. The following documentation is part of the SDLC package that is maintained throughout the software lifecycle:
A. Validation Documentation
a) Validation Plan
A Validation Plan is a high-level document that defines the overall validation strategy for an organization’s system. An organization can have a Master Validation Plan as an overarching document to multiple validation plans. A Validation Plan should be created per system (eg, each EDC system, Quality Management System such as Trial Master File (TMF), Training, etc.) and may include components of the IQ, OQ and PQ phases. A Validation Plan may also be created for each phase per system.
A Validation Plan documents and describes the system being validated; the environment in which it is installed; assumptions and limitations of the project; the testing and acceptance criteria that will be in place; the standard operating procedures to follow; and the roles and responsibilities of the validation team.
Per GAMP 5,6 the Validation Plan should be a brief document but should at least cover the following topics:
“Introduction and Scope
System Overview
Organizational Structure
Quality Risk Management
Validation Strategy
Deliverables
Acceptance Criteria
Change Control
Standard Operating Procedures
Supporting Processes
Glossary”6
b) Requirements
A requirement outlines a condition that needs to be in place for the software to perform the way it is expected.
Per GAMP 5: “The requirements should define clearly and precisely what the system should do and state any constraints. Requirements should be reviewed and approved.”6
Requirements may include the following:
Business Requirements Specifications (BRS) – expectation of processes in standard operating procedures (SOPs)
Infrastructure requirements – the necessary staff, facility and equipment
Functional Requirements Specifications (FRS) – including system functionality, performance and security
User Requirements Specifications (URS) – user interaction within the system with specific functionality.
Documented requirements should be clear, concise, specific, and unambiguous. For example, a requirement related to security may meet the regulatory needs for 21CFR Part 112 as well as an organization’s SOPs (2011 Von Culin).10 A requirements document can address both needs – regulatory and SOPs (refer to Appendix A).
Throughout the lifecycle of the software, FRS/URS should be reviewed to address any changes that occur based on new features during software upgrades or patches. Updates to SOPs may impact the URS and should also be evaluated in case requirements are to be added/removed. The URS document should be a controlled document.
c) Test Plan
A Test Plan is a detailed document that defines the test strategy, objectives, schedule, estimation, deliverables, and resources required to perform testing for a software product. A Test Plan is put in place to make sure that the product conforms with the requirements. Per the GAMP 5.0,6 the Test Plan may contain the following components:
“Which types of testing are required
The number and purpose of test specifications
The use of existing supplier documentation in accordance with the results of the supplier assessment
Test phases
The approach of supporting test evidence
Procedures for mapping test failures
Format of test documentation
Use of test metrics”6
The information from the different components of the test plan may be combined into one test plan per the organization.
Acceptance criteria may need to be repeated in the test plan, especially if there are some variations from the validation plan acceptance criteria.
Once a test plan has been established, the testing may be split per test case/test procedure. The testing strategy may look like Figure 2:
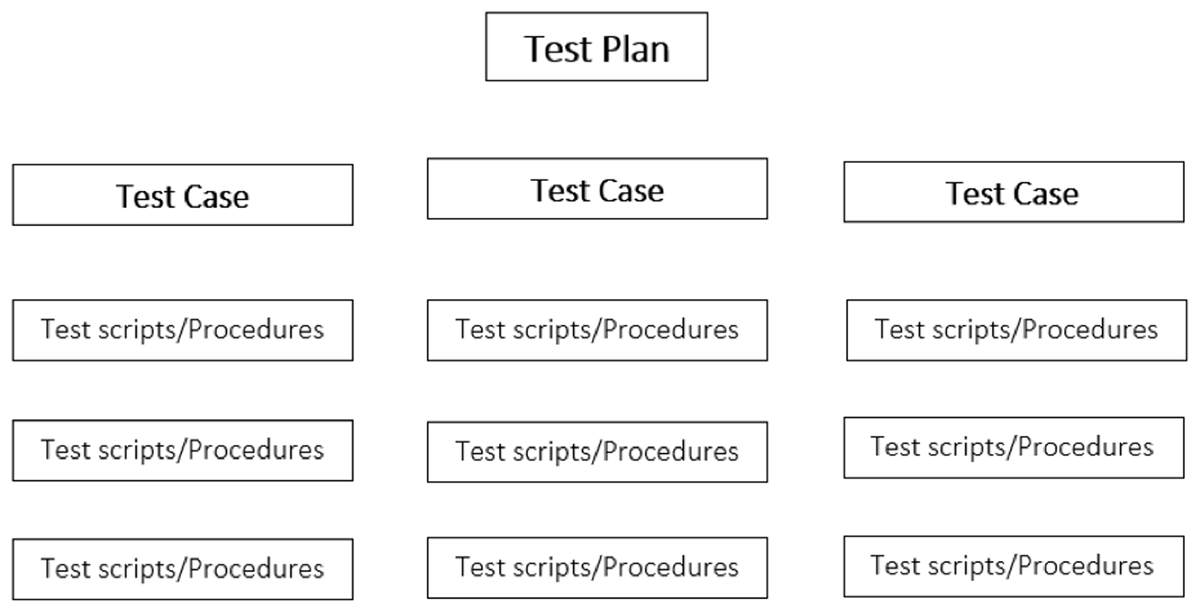
Example of Testing Strategy.
d) Test Cases
What is the difference between a test case and test script/procedure?
Per IEEE std 829-2008, Section 3.1, item 3.1.41, a test case is defined as “A set of test inputs, execution conditions, and expected results developed for a particular objective, such as to exercise a particular program path or to verify compliance with a specific requirement” and a test procedure (Item 3.1.50) is defined as “Detailed instructions for the setup, execution, and evaluation of results for a given test case.”13
The validation team should decide how the testing documentation will be organized. The use of a test case may make it a bit easier to organize the testing by requirement categories or by a set of scripts that have to run together due to business requirements or because the set of steps have to be run sequentially. Testing can also be conducted at a script level. The goal is to ensure that all FRS/URS are tested thoroughly and to document the outcome status of each of the scripts/procedures or test cases.
The challenge of a test case is that the status of Pass/Fail can only be obtained if all the scripts underneath have a Pass status. It may not always be convenient to group too many test scripts together, depending on the complexity of the requirements.
The tests need to be conducted per the test specification and the testing evidence needs to be well documented to show that the software meets the requirements (refer to Appendix B).
e) Tools/Reports
Requirements Traceability Matrix – A tool used to trace and document the requirements with the test cases. A Traceability Matrix is an important document that allows the validation team to confirm each requirement has been tested. It serves as an overview of the validation effort (refer to Appendices C and D).
-
Testing Summary Report – The test summary report is a document that provides a summary of the testing objectives, activities, and results based on the test plan.
An example of a standard table of contents for a testing summary report is given below.
Table of Contents:
1. Introduction
2. Overview
3. Summary Assessment
4. Detailed Test Results
4.1. <Test Category/Function>
4.2. <Test Category/Function>
5. Variances
6. Test Incidents
6.1. Resolved Test Incidents
6.2. Unresolved Test Incidents
7. Recommendations
Appendix A: Test Incident Reports (TIRs)
Appendix B: Record of Changes
Appendix C: Glossary
Appendix D: Referenced Documents
Appendix E: Approvals
Appendix F: Notes to the Author/Template Instructions
List of Figures
List of Tables
Table 1 – Test Case Summary Results
Table 2 – Test Incident Summary Results
Table 3 – <Test Category/Function> Results
Table 4 – Example Test Failure Report (TIR)
Table 5 – Failure Description
Table 6 – Failure Resolution
Table 7 – Record of Changes
Table 8 – Glossary
Table 9 – Referenced Documents
Table 10 – Approvals
Software Verification and Validation Summary Report – A summary report is a document that contains the scope, test cases, deviations and how they were resolved, and confirmation to show that the system(s) meet the requirements of the overall project. Approval of this document consists of the actual release of the software into production. (GAMP Good Practice Guide Testing of GxP systems, Appendix T7)6
An example of a standard table of contents for a validation summary report is given below.
Table of Contents:
1 SUMMARY REPORT INTRODUCTION
1.1. Objectives
1.2. Scope
1.3. Assumptions
1.4. Exclusions
2 ACRONYMS AND REFERENCES
2.1. Acronyms and Definitions
2.2. References
3 TESTING SUMMARY
3.1. General
3.2. Documentation
3.3. Equipment/Materials
3.4. Testing Exclusions
4 RESULTS OF VALIDATION TESTING
4.1. Installation Qualification Tests
4.2. Approval to Proceed to Operational Qualification
4.3. Operational Qualification Tests
4.4 Performance Qualification Tests
5. CONCLUSION
B. Security/Access Control
System security is critical to maintain authenticity, confidentiality, data integrity, and data quality. Every sponsor/CRO/vendor must have procedures in place to grant access to the GxP system to authorized personnel only and protect against willful threats or changes. In these procedures, the criticality level of any breach of security must be illustrated. (GAMP 5 – Appendix O11)6
There are two types of access: physical access and logical access.
Physical access refers to the space in which the GxP system equipment resides. As stated in 45 CFR Part 164, Section 164.310 “Standard: Facility access controls. Implement policies and procedures to limit physical access to its electronic information systems and the facilities in which they are housed, while ensuring that properly authorized access is allowed.”12 The physical access should only be granted to trained professionals, and the list of authorized personnel should be readily available for audit purposes.
For example, the server room may reside on-site or off-site. Controlling access to an on-site server room may be easier as the authorized personnel would also be on site and can take immediate action in case of a breach. For off-site access, the sponsor must ensure that the subcontracted entity has procedures in place and can be audited regularly. The sponsor must be notified immediately in case of a breach.
Different types of access control can be used (eg, biometrics, access cards/fobs, access codes, keys, etc.). These should be evaluated to ensure appropriate controls are in place based on the space and who may be granted access and checked regularly by the authorized personnel to avoid any potential breach. A log, such as the one in Figure 3, can be used to record the reason for actual entry to the server room on top of any electronic system that may already be in place:
Logical access refers to the remote access to the system used for identification of the user(s). Only authorized personnel should be granted access to the software. Technical safeguards should also be assessed to ensure only authorized personnel are granted access (45 CFR Part 164 – section 164.312).12 An organization should also assess if Single Sign On or Multi-Factor Authentication is required. This functionality allows less vulnerability to the system(s).

Example of Risk Class.
Usually, the EDC software access is granted by role (eg, admin, CDM, site personnel, monitor). The base role access protects the data from unauthorized personnel, so data integrity and quality are maintained.
Each role created should be documented and list all the tasks that are allowed within the role.
It is the organization’s responsibility to set a password policy to ensure proper system security and be compliant with regulations. General information can be found in multiple guidelines:
21 CFR Part 11(section 11.300)2
ICH GCP E6 (section 5.5.3 addendum)3
National Institute of Standards and Technology (NIST) Special Publication 800-63 – Digital Identity Guidelines Section 5.4 – Risk Assessment and Compensation Controls “Agencies SHOULD implement identity services per the requirement in these guidelines and SHOULD consider additional techniques and technologies to further secure and privacy-enhance their services.”14
GAMP 5 section 4.3.76
45 CFR Part 164 Section 164.308 Administrative Safeguards
As indicated in the NIST special publication, each organization should assess their security risks and employ the appropriate measures to restrict access and maintain control. For example, some of the common passwords requirements may be:
A temporary password reset by the user after initial login
A renewal policy at specific intervals (eg, 30, 60, 90 days)
Specific system requirements for passwords may also be applied (eg, numeric, special character, capitals, length of password).
Each user must have a unique account credential that will not be shared with any other personnel.
Any incident to physical or logical access must be documented and reported to the appropriate personnel. Procedures should define who is to be notified of the incident based on the criticality level (eg, system owner vs IT). Each incident should be investigated and Corrective and/or Preventative Action(s) (CAPA) applied.
Logical access does not only apply to systems but also to files exchanged between organizations, specifically if these files contain organized data structure (eg, Excel files). A setup of file transfer protocol (FTP) should be put in place to ensure access to this information is restricted to only the authorized personnel. If a FTP cannot be used, files may be sent via email with password protection. This process would avoid security risks of files being sent or intercepted by malicious users. The password must not be shared in the same email as the file attachment.
C. Change Control/Maintenance
a) Documentation
Once the system is validated and released for use, it must be maintained and kept in a validated state during its operation. As mentioned in Section 4 of GAMP 5,6 an important step of the “Operation” Life Cycle Phase is to maintain documentation that defines the procedures and steps to oversee system maintenance and change control. Furthermore, ICH E6(R2)3 describes computerized system topics that should be covered in SOPs, which include system maintenance and change control.
b) Change Identification
Software changes may be identified through different routes, including periodic review, process improvement initiatives, changes to requirements (eg, changes to the clinical study or to regulations), and incident management.
Periodic Review: GAMP 5 (section 4.3.5) recommends periodic reviews to ensure that the system “remain compliant with regulatory requirements, fit for intended use, and meet company policies and procedures.”6
Process improvement: Periodic reviews and user feedback may provide opportunities for software improvement.
Requirement change: To maintain system integrity, the system must stay in compliance when requirements change during its operation (eg, change to clinical study or regulatory requirements).
Incident management: Performance and other unplanned issues should be resolved through an incident management process that identifies, evaluates, and tracks resolution. Some incidents may be resolved through the Corrective Action Preventive Action (CAPA) process that is important for preventing recurrence of issues, especially high-risk issues.
c) Evaluation
All software changes should undergo a formal change control process. This process begins with an evaluation of the change that assesses impact to the existing system and the risk and complexity of implementation (intended and unintended impact). An important aspect of change analysis is to identify the root-cause of the issue and to assess the need for resolution based on the impact to system/data quality and patient safety. System security is an example of a high-impact issue. The evaluation of the change should be documented and referenced throughout the change implementation.
d) Implementation
If the change is deemed necessary after careful evaluation and authorization, it must then be tested and documented. Examples of change implementation are software upgrades and security and performance patches. As noted in the FDA Guidance General Principles of Software Validation,7 the validation of the software must be re-established no matter how large or complex the change is. The testing should not only cover the individual change but should also determine if the change impacts previously validated requirements. This step, known as regression testing, can be performed by rerunning tests that had previously passed to verify if the results are the same. Regression testing is used to demonstrate and provide confidence that the system has not been negatively impacted by the change. It is recommended to describe the approach for regression testing in the test plan.
D. Training
Training is an important requirement of Good Clinical Practice ICH E6.3 An individual should be qualified to perform his/her duties based on education, experience, and/or specific training(s) (ICH GCP E6 R2 Section 2.8).3 For an individual to participate in the software validation activities, they should have received training on 21 CFR Part 11 as well as the organization’s procedures related to software validation. Training should be targeted based on the role the individual is to play in the SDLC and validation.
A training log can be included either as part of the software documentation package or within the individuals’ training records. These training records must be readily available for audit purposes.
Before deployment of the software to users, a training plan should be set in place by user role. The training materials are developed to include the activities the user is expected to perform within the system. The user training and user manuals should be accessible per role. No system access is to be granted until completion of the training is verified. At the time of any update to the software, training materials should be reviewed and assessed for adjustments. If changes are applied, new training materials should be deployed and documented to keep the users qualified to use the system.
In terms of what the training may look like, it will be dependent on the organization’s procedures, and specific usage of the system should be part of the training/user manuals. The training delivery method is to be determined as to provide the best and most efficient approach (either in person or online) to train users. The ultimate goal for an organization is to provide training to each user so they are qualified to interact with the system based on their role (eg, a site coordinator must know how to perform data entry, an investigator may only need to sign off in the system, a data manager needs to be able to review and query the data).
E. Backup/Recovery and Business Continuity
a) Backup and Recovery
Backup is the process of copying records from one system to another to protect against the loss of data. Recovery is the process to restore the records from a backup copy.
An organization should have procedures in place to describe how the system will be backed up and recovered (GAMP 5.0 Appendix O9 section 4.4).6 The procedures should cover all details, including frequency, type of backup used, storage on-site vs off-site, number of backup copies, retrieval, rotation of media, and retention.
It is the organization’s responsibility to make sure that the procedures in place are adequate based on needs and risk. These procedures should be tested thoroughly and are usually referred to as Disaster Recovery Plan or Business Continuity Plan. The frequency of testing the backup and recovery procedures may also be included in a Periodic Review document.
In general, a system backup consists of either a full backup, incremental backup, or a combination of the two. As mentioned earlier, the frequency for each type (eg, daily, weekly, monthly) is to be described in procedures that may be determined by the business or application owner in conjunction with the Information Technology (IT) department.
A full backup is performed when a system is “offline” and contains every file on the system.
An incremental backup is typically performed while a system is running and includes only those files that have changed since the last full backup.
System backups are performed on a periodic schedule as determined by business or application owners in conjunction with IT Services.
A suitable backup format (eg, disk or tape) is determined based on the system/hardware requirements and procedures. The backup media can be overwritten as defined in procedures. The backup format used should also be thoroughly tested as it may become corrupt and unusable for recovery.
System backups are typically stored on both disk and tape media. Once the backup retention period expires, the disk and tape media are either re-used (overwritten), erased, or destroyed in an approved manner. At a minimum, backups consist of two copies. The first copy is stored and located on a disk pool located in the data center. The second copy is stored on tapes that are sent off-site as defined in the organization’s IT SOPs. Both copies have the same retention settings, which are specified in the System Backup Profiles.
Any failures that occur during a backup should be investigated and documented, and proper actions should be taken (eg, restart of backup, new media to be used, etc.).
Note that off-site storage facilities should be secured in the same manner as the system itself. It is the sponsor’s responsibility to review the off-site procedures as well as perform regular audit(s).
If recovery is to occur, the IT team discusses the strategy with the application owner. The risks associated with restoring a backup should be assessed. The restoration could have an impact on the data and system. There may be some system downtime and users should be contacted to inform them that the system is not available. Additionally, some data may need to be re-entered by the user.
b) Business Continuity
A Business Continuity Plan (BCP) is a documented set of processes that would restore the business activities after a disruption. The plan usually encompasses the critical business activities while restoring technical systems. The technical systems are handled by the IT department and are usually referenced as a Disaster Recovery Plan (DRP).
The BCP should cover all departments/levels of an organization and describe the processes that will be put in place in case a disaster occurs. (GAMP 5 – Appendix O10 -section 1)6 All types of disasters should be assessed when developing a BCP as well as the level of disruption:
Physical disaster
a remote worker is localized in a disaster area
an office is not accessible
multiple offices are not accessible
Logical disaster
a computer is corrupted
a network service is not accessible
all systems are down
The goal of a BCP is to allow critical activities to be restored as soon as possible after a disaster: a priority of critical functions should be listed and reviewed periodically by the business/application owners in collaboration with the IT department.
At the time of the disaster, the priority list will be used to restore the activities/systems in order of criticality defined in the BCP. It is therefore important to assess all potential risks for the organization, including the use of third-party organizations in the day-to-day activities (eg, off-site EDC facilities, off-site backup).
In the BCP, the responsible person(s) who will enact the plan should be listed in the primary contact order. The BCP should also be communicated throughout the organization and tested regularly as business criticality may change over time, equipment may be updated, or software changed.
If the sponsor is using a third-party vendor for any data collection, it is important that the BCP of that organization is reviewed and meets the minimum requirement of its own BCP. The third-party vendor should also review and test their BCP regularly.
Here are examples of sections that should be present in a BCP:
List of the team members’ part of enacting the BCP
List of the organization’s critical activities
List of sites and evacuation plans
Type of potential disasters
Severity of the disasters
System criticality and expected recovery time
Communication path internally to staff and externally to sponsor/vendors
ISO 22313:2020 “Security and resilience – Business continuity management systems – guidance on the use of ISO22301” can provide some further guidance for creating a BCP.15
F. Decommissioning/Retirement of System
Systems eventually end the journey in their lifecycle; the process is called decommissioning/retirement. Decommissioning a system is different from simply removing user access to a system or to a study database at the completion of a clinical study. System retirement completes the lifecycle of an entire application, with the intention that the system will no longer be available for future use.
Validation of computerized systems plays a vital role in decommissioning of the system in terms of accuracy, reliability, and consistent intended performance. Before a system is decommissioned, a decommission plan should be established by the various key business partners (eg, process/system owner, IT, and QA).
It is recommended that the time of decommissioning takes into consideration whether or not the clinical study will be used in the terms of a marketing authorization application in the near future, in which case it could be recommended to keep the database(s) accessible.
A dated and certified copy of the database(s) and data should be archived and available on request during the data retention period set by law or regulatory requirements.
If the sponsor is using a third-party vendor, the sponsor should ensure system decommission is addressed in contract and archived formats are available to provide the possibility to restore the database(s). Clear requirements from the sponsor should be addressed with the third-party vendor before the system is decommissioned (eg, audit trail availability, file formats, system metadata). This includes the restoring of dynamic functionality and all relevant metadata (eg, audit trail, event guideline, implemented edit checks, queries, user logs, etc.).
Where recommissioning is no longer possible, the sponsor should ensure that all the data including metadata files (eg, audit trails) is available in dynamic data files. The sponsor should review the system to determine the audit trails and logs available in the system and how these would be retained as dynamic files (eg, spreadsheets with automatic calculations), include automatic processing, and/or enable an interactive relationship with the user to change content (eg, eCRF). A certified electronic copy may be retained in different electronic file formats to the original record, but the equivalent dynamic nature (including metadata) of the original record should be retained. Where a third party is involved, the process for system recommissioning should be addressed in the contractual arrangements. Static formats (eg, PDF scans containing information or data that are fixed/frozen and allowing no change in the content) of dynamic data will not be considered adequate.
As per GAMP 5 System Retirement Planning Appendix,6 the system retirement process is documented in a system retirement plan which should receive input from all relevant functions. “Inputs to the planning process may include:
record retention and destruction requirements for historic data or records
identification of the current software and hardware configuration as well as interfaced systems, equipment or instruments
identification of any external systems that rely on data or records from the system.
The extent and rigor of planning should be based on the system impact and risks associated with loss of data.
The System Retirement Plan typically should be approved by the process owner and Quality Unit, and others as required, such as the system owner. The System Requirement Plan should describe the approach to be undertaken, including:
introduction
roles and responsibilities
overview and implications
business process description
retirement approach
data and record migration, archiving and destruction
verification approach
ending system maintenance and support
change management
schedule
retirement execution
system documentation”6
7) Risk-Based Considerations for SDLC
Risk management is a systematic process for assessment, control, communication, and review of risks to the quality of the software across the software lifecycle. Risk management is an essential aspect in every software development process. This process involves multiple phases, each phase being vulnerable to different types of risk factors. Identifying, understanding, and documenting these risks at an early stage is very important in managing the risks.
There are different kinds of risks in SDLC:
Plan risk: Plan-related risk refers to time-related risks or project plan-related risks. These risks mainly indicate project activity running behind schedule, resulting in a delay of the software delivery. Some of the reasons for plan risk are improper assessment of estimated time required for each activity, inefficient tracking of project status, and improper resource allocation. If plan risks are not handled properly, a delay in software delivery may occur, eventually impacting the organization.
Budget risk: Budget risk refers to money-related risks, which occur when a software development project exceeds budget limits. Financial aspects of the project should be managed per the original agreement. If the finances of the project are not managed, budget-related risks may arise. Some of the reasons for budget risk are incorrect estimation of budget, finances of the project not being tracked properly, and unexpected changes in the project not considered from a budget perspective.
Operational risk: Operational risk refers to risks that occur in operational activities during software development. Some reasons for operational risk include insufficient number of skilled resources, insufficient training, and attrition.
Technical risk: Technical risk refers to the risk associated with the functioning or performance of the software. Some reasons for technical risk are inappropriately written software specifications, frequent updates in the requirements of software, and underestimated complexity in software development.
After categorizing the risks, the next step in a risk-based approach is to define the steps involved in risk management. Risk management could be broadly categorized into the following:
Identification of risks
Evaluation of risks and their impact
Identification and implementation of controls
Review of risks and monitoring controls.
a) Identification of risks
Initial risk assessment should be based on the various steps followed during software development, business process, user requirements, regulatory requirements, and known functional areas. Based on the initial risk assessment and the impact on the software, an assessment of whether or not the risk is acceptable needs to be conducted. If the risk is an acceptable level, then the subsequent steps in risk management would not be required. Known risks and their mitigation plan should also be considered during identification of risks.
b) Evaluation of risks and its impact
Once the risk is identified, it needs to be further evaluated in terms of the degree of adverse impact, frequency of occurrence, and how to prevent the negative impact due to risk from impacting the software development. Impact to the business and to any other interfacing systems should be considered.
Risks affecting various aspects of software development could be assessed using one of the following methods:
-
Severity of impact is plotted against the probability of risk to occur, giving a Risk Class (Risk Class = Severity × Probability). Severity is assessed in terms of the impact on patient safety, product quality and data integrity. Probability is defined as the likelihood of the issue occurring. The Risk Class is then used to determine the Risk Priority (Risk Class x Detectability) by assigning the detectability or the likelihood that the issue will be identified before a bad outcome occurs.
Example: Access restrictions are not working as intended in the system. In this case, severity would be high as there is an impact on data integrity. However, the probability of this happening is low so it will be categorized as Risk Class 2. The validation team then determines that the detectability is low because it is likely to go unnoticed prior to a security breach. Therefore, a Risk Class of 2 with low detectability results in a High Risk Priority which may require additional validation scripts to be run (refer to Figures 3 and 4).
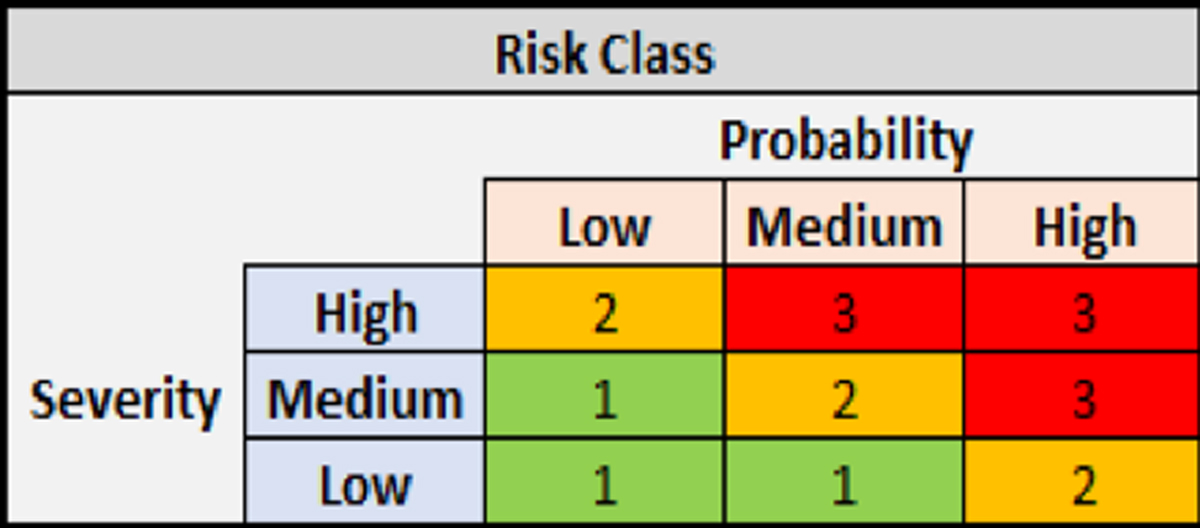 Figure 3
Figure 3Example of Risk Class.
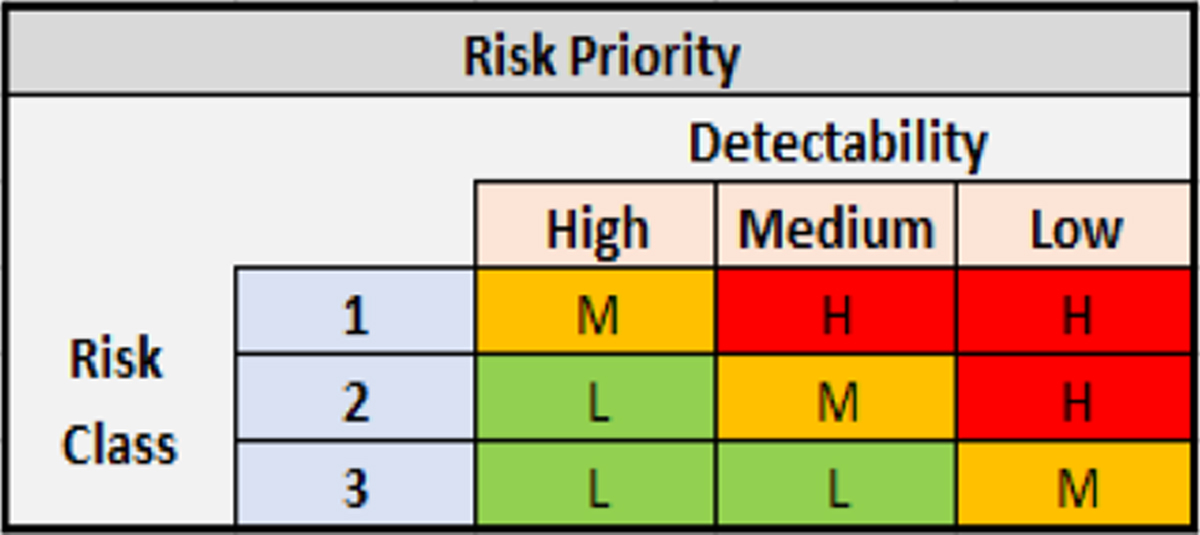 Figure 4
Figure 4Example of Risk Priority.
-
Another way of risk assessment has been described by Von Culin 201110 which is as follows:
In this method, questions around the various factors that would help in determining system risk are asked and the responses are evaluated for risk and given a weighting (see Figure 5).
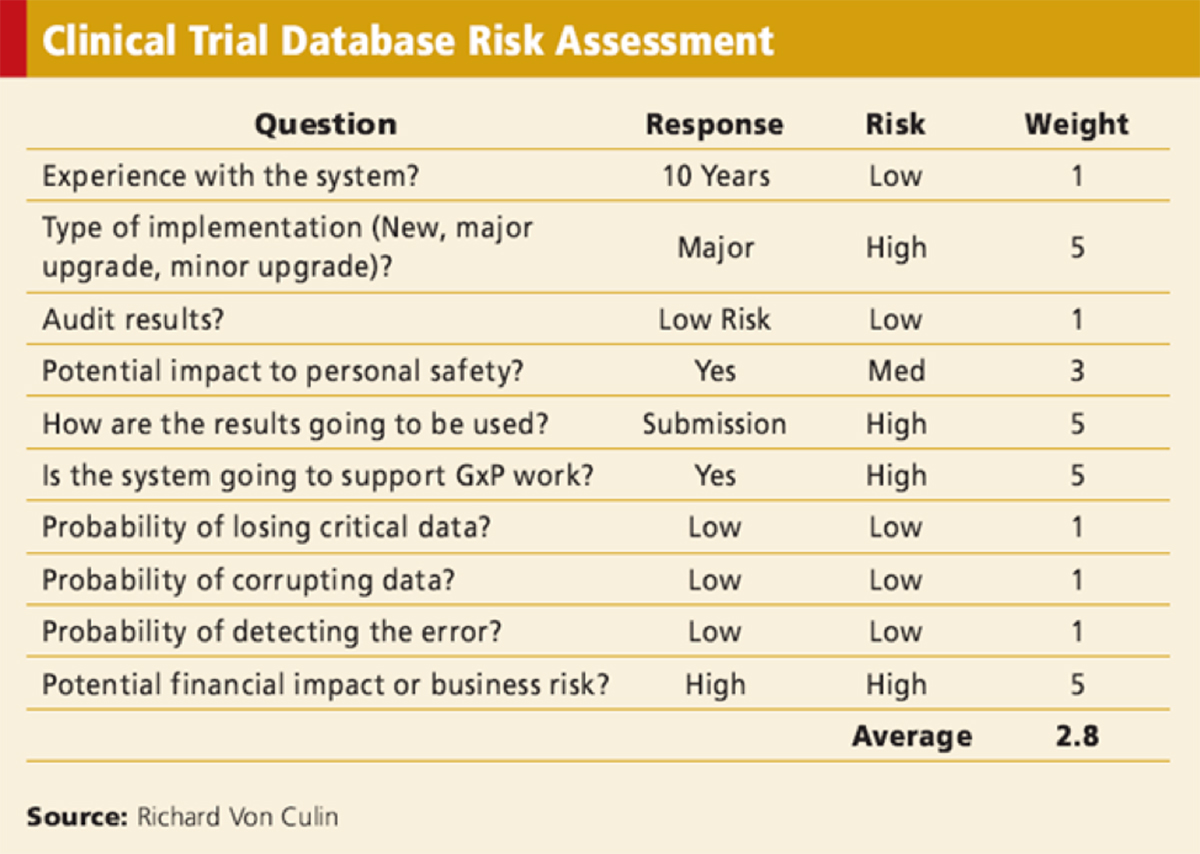 Figure 5
Figure 5Clinical Trial Database Risk Assessment.
There are a number of different tools for assessing risk, including flowcharts, check sheets, process maps, and cause and effect diagrams. For some of the common techniques, see Annex I: Risk Management Methods and Tools of ICH Quality Risk Management Q9.16
c) Identification and implementation of controls
Controls are measures put in place to reduce risk. Controls could be part of the software, SOP, or could be present downstream to identify the issues in software after they have occurred. An example is Quality Control (QC) of data once data are extracted from the Clinical Data Management System (CDMS). In this step, data are reviewed to check whether their format is correct and are extracted per the specifications provided for the system, built or not. If there are any issues identified in the data, then how the system should be fixed must be assessed. The aim of controls is to eliminate risk through process update or software reprogramming, reducing probability of occurrence of issues in software, adding system checks to detect issues at the failure stage itself, and adding checks in downstream processes to identify an issue once it occurs.
Controls in software development should be automated within the software and should work in real time for immediate identification of issues. Such controls should be independent to avoid failure of checks in place. Examples of controls to reduce risk include automated data verification checks in software and/or replicating the production environment of CDMS in a test environment.
Risks that cannot be reduced with the help of controls must be assessed further to manage them through system redesign and making changes in the process of software development.
d) Review of risks and monitoring controls
Developed software needs to be monitored periodically or at a defined stage in the process to keep the risks in check. Review would involve ensuring all the controls implemented are working fine and also to assess if there are any unknown risks that should be identified and mitigated.
Results of risk evaluation should be documented as part of the risk management process. If any changes are required in the risk management process, those should be agreed upon by members involved in the risk management process and documented appropriately. Frequency and scope of controls review would depend on the level of risk.
8) Vendor Qualification and Oversight for Licensed Software
Vendor qualification is the process by which a vendor is evaluated by the sponsor to determine if the vendor qualifies to provide the products or service that the sponsor requires and can manage the risks effectively. Developing and implementing a vendor qualification program is an important step in ensuring compliance with the ICH-GCP regulations.
According to ICH E6(R2), sections 5.2.1 and 5.5.3.a, respectively, “the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor”3 and “the sponsor should ensure and document that the electronic data processing system(s) conforms to the sponsor’s established requirements for completeness, accuracy, reliability, and consistent intended performance (ie, validation).”3 The sponsor is ultimately responsible for the validation of the clinical study processes that are supported by electronic systems and is also responsible for providing sufficiently documented evidence to GCP inspectors on the validation process and on the qualification of the electronic systems.
To implement the vendor qualification and auditing program, the sponsor company needs to have appropriate written procedures. Specific documentation for use of auditors needs to be in place.
a) Vendor Audit Checklist
The purpose of the Vendor Audit Checklist is to ensure that:
Data collected in the system is not corrupted or lost
Data is secure, with appropriate access authorizations
Approvals cannot be rejected
Changes to data can be traced
Attempts to falsify records are made difficult and can be detected.
A Vendor Audit Checklist could include the following:
Product history and development plans
Methods of assuring quality, Quality Management practices, metrics collected, staff qualifications, use of subcontractors, awareness of regulatory requirements
Documentation to demonstrate that system access is limited to authorized users
Tools, practices, and conformance to SDLC used
Requirement specifications, traceability, reviews and approvals, accuracy, and conformance to process
Programming language standards, version control, builds, tools, code reviews
Testing strategy, specifications, scripts, procedures, completeness, remediation, independence of testers, traceability to specifications
Documentation of release, archiving of tools release and documentation
Explanation of services, fault reporting and resolution
Documentation management, software configuration management, change control, security, and conformance to process.
Please refer to GAMP 5 Section 9.1.26 for items that can be included in a Vendor Audit Checklist.
After the vendor is qualified and performs the SDLC tasks per the Scope of Work (SOW), the sponsor is responsible for oversight. The total involvement and approach to oversight should depend on the risk of the task. For example, the sponsor may be involved in User Acceptance Testing (UAT) to ensure that the outsourced data management software meets the intended use of the sponsor and that it conforms to regulations. It is likely not necessary or feasible to perform UAT on all system components. Instead, it is more efficient to perform a risk-assessment and perform UAT on only critical components that have the highest impact on the data with consultation from data management and a biostatistician (2016 Peterson)17. Even if the vendor provided evidence for 21 CFR, Part 11 during qualification, it is recommended that the sponsor perform UAT or ask for evidence of compliance during or after the system is validated. For more information on vendor oversight, refer to the Vendor Selection and Management Article in the Good Clinical Data Management Practices (GCDMP).
b) Validation Documentation
The vendor assesses all the documentation listed in Section 7 to ensure that it meets the 21 CFR Part 11 requirements, as well as sponsor’s QMS requirements. Also, as defined in Section 7, IQ/OQ/PQ/UAT phases need to be conducted.
9) SDLC for In-House Systems
An organization may develop its own software to meet its business needs. This is referred to as an in-house system. In regard to the use of in-house systems in clinical studies, all of the SDLC components (as described in section 7) apply. The system owner should exercise due diligence and rigor, with emphasized responsibility for all aspects of the validation, implementation and maintenance of the software, as these are critical to ensure its functionality and the reliability of the data collected.
Here are some additional tasks to include when planning to utilize an in-house EDC system:
In designing the in-house system, prepare for the following:
user-friendly data entry and reporting tools
data security, including user access and appropriate permissions based on roles
data edit checks
audit trail (and audit trail reviews)
adverse event and medication dictionary coding
electronic signatures
randomization of study treatments
data output formats for analyses (eg, SAS, CDISC/Study Data Tabulation Model [SDTM]-ready datasets) as well as for archival
database locking
Notify the internal organization of the in-house software, as the company’s information and privacy policies may require further vetting/registration
Plan for method of users’ connectivity to the in-house system (eg, Remote Desktop Protocol [RDP], Application Programing Interface [API], dedicated server, cloud, stand-alone computers, cell phone applications)
Have a team to oversee the operation, maintenance, change control, and periodic reviews of the system
Ensure SOPs are written to cover all processes involved in utilizing an in-house system
Create technical/process manual(s) and training materials for internal and external users of the in-house system
Plan for technical support to be available to the clinical site users
Provide a software validation certificate to the clinical site(s); this could be accessible within the in-house system itself
(10) Utilizing Open-Source Software
Open-source refers to code available to everyone and allows users to freely modify. Validation of open-source software (OSS) poses a challenge because there is no predefined vendor and the OSS user needs to adopt a risk-based validation strategy to meet regulatory requirements for computer system validation. The basic principle is to adhere to testing, validation, and documentation. According to Eudralex, annex 11 computerized systems, “Risk management should be applied throughout the lifecycle of the computerised system considering patient safety, data integrity and product quality. As part of a risk management system, decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment of the computerized system.”18
Utilizing open-source brings cost effectiveness, flexibility, customization, and easy-to-use methodology. There is no fear of being locked in with a specific vendor or CRO and developing a blueprint based on the business need. In OSS, the system should focus on both physical and logical architecture, associated data flows and interfaces with other systems or processes if applicable, and security standards. Although a community developer supports OSS, an internal cross functional approach is required to achieve a milestone that includes Business Owner, System Owner, System administrator, QA, and IT. The responsible user should ensure that the system has been developed in accordance with the company’s quality management system (QMS) and compliant with 21 CFR Part 11, GCP and HIPAA.2, 3, 19
a) Validation Documentation
Software used in clinical studies should be validated regardless of the licensing agreement for that software. Like any software validation, OSS also requires validation for the three stages (IQ, OQ, PQ) when responsible for hosting and maintaining the software (refer to Section 7).
The URS and functional specifications should be clearly defined and documented. They are defined as individual items and each such item can be individually tested based on the system validation plan.
b) Tools/Reports
Software should be developed in accordance with an organization’s QMS.
Develop SOPs for software development, system administration, validation, change management, and security models.
The basic tools include operating system, application server, and database to configure or install any OSS should be present.
Testing Management and/or Testing Automation Systems may be applied to centralize and thereby better manage system validation activities. These types of tools enable requirements gathering, management, and execution of test cases, defects logs, and the generation of different types of reports, such as user requirements, test scripts, evidence reports, and requirements traceability matrix.
11) Study-Specific Customization of Software
Once software (eg, clinical data management system or CDMS) has been validated, study-specific configuration is tested to demonstrate that the requirements for the study implementation have been documented, developed and tested for its intended purpose. The FDA’s guidance on computerized systems used in clinical investigations states that “each specific study protocol should identify each step at which a computerized system will be used to create, modify, maintain, archive, retrieve, or transmit source data.”8
a) Validation Documentation
Study-specific programming may vary greatly in complexity. An example of a more complex logic is programming that uses multiple conditions or “if statements”. More complicated logic should be validated by a user who is well-versed with programming. In addition, FDA’s guidance on general principles of software validation states that “an independent evaluation is always better, especially for higher risk applications”7. This may involve the validator programming the same logic on their own and verifying that the results match.
It is beneficial to be consistent and implement standards for reuse when possible to decrease overall validation efforts. Consistent and thorough test logs will limit the risk of errors in configuration. Standard configuration that is tested for one study could be reused for another study without retesting if the underlying code has not changed.
b) Database Testing
Validating database configuration is essential for having confidence in the integrity and accuracy of study results. Per ICH requirements (ICH E6 R2 5.5.3), “the approach to validation should be based on a risk assessment that takes into consideration the intended use of the system and the potential of the system to affect human subject protection and reliability of trial results.”3 Study-specific validation of a database can be summarized by three major categories: database configuration (eg, case report form settings, visit setup, security, etc.), data entry or capture, and other study-specific programming (eg, edit checks, notifications, randomization schemes, etc.).
1) Database configuration
An important step of database configuration is to ensure that the required data are expected and retrieved when necessary. Case report form (CRF) entry should align with the protocol schedule and prevent data capture that is not required per the protocol (eg, unscheduled visits). If a CRF is not available when expected, it could be detrimental to study results if analysis data are missing as a result.
Another example of a database configuration test is verifying that a CRF is configured to allow the investigator to sign the CRF after all queries have been addressed. One check may be to ensure the tester cannot sign the form when there is an open query. Another consideration is to confirm that the study protocol requires a signature on the data and that the open-query restriction meets the needs of the end user.
In addition, it is crucial that users see only the data that is relevant for their role. Title 21 CFR Part 11, Section 11.10 states the following regarding authorized access:
“Limiting system access to authorized individuals
Use of authority checks to ensure that only authorized individuals can use the system, electronically sign a record, access the operation or computer system input or output device, alter a record, or perform the operation at hand”2
For example, a test may confirm that an investigational site user can only see data for their site and that, if applicable, they cannot see data that they are blinded to. Security setup and preventing bias by ensuring that a blind is maintained when applicable to a study design is critical to the integrity of the study results.
2) Data entry or capture
When testing a study’s data capture, one of the most important considerations is to ensure that data entered through a data entry screen or captured via some other transfer process (eg, electronic lab data transfers) map to the correct variables and that the data are stored in a way that can be used for its intended downstream purpose (eg, primary analysis).
The tester may enter test or “dummy” data and verify that it is stored with the appropriate variables. Testing may be performed on all data, regardless of whether the data meets defined data structures. Test cases may include data that does not meet the data definition for length and/or type (eg, 11 characters in a field that accepts 10 characters) to ensure it is stored appropriately by the system. The tester may confirm that variable lengths are sufficient to prevent truncation or rounding and that character and numeric formats provide the necessary output for the analysis file. Test cases may also vary enough to have confidence in the data mapping (eg, do not enter all test data with a response of “No” on a CRF that includes consecutive Yes/No field types).
For data transfers, test data transfer files so that output and data extracted from the database can be reviewed to ensure that the variables were correctly added and saved within the database structure. Some studies may derive data from other fields (eg, Body Mass Index (BMI) derived from height and weight). It is important to test all components of the calculation or derived value, especially if the field is used in analysis. The tester may think about how the data are impacted if related data changes. For example, a BMI value may update if height is updated after the CRF is saved.
3) Other study-specific programming
Software is becoming more complex with the addition of new capabilities. Some of the common capabilities include notifications that trigger or alert users when a certain condition is met (eg, entry of an adverse event), randomization modules, data loading or transfer programming (eg, loading adverse event coding variables or loading central lab data), and programming written to validate the data (eg, edit checks, out-of-range checks, etc.). For example, if the database is programmed to flag out-of-range data, one validation step would be to ensure that flags appropriately trigger upon receipt of the data. It is recommended to use a risk-based approach when determining validation efforts for study-specific programming. For example, it is recommended to validate programming if it impacts the quality of the data or is used to make decisions that impact the analysis or the submission data.
4) Other database validation considerations
Database entry/capture validation testing may help to identify key entry management issues. For example, the database may not accept duplicate entries. Additionally, the system may not allow data unless the unique identification variables (eg, primary/secondary key variables) are provided. The audit trail for the study should be validated and protected so that all manipulations of the study database or external files are documented by date, time, and user stamps in an unalterable audit trail that can be accessed throughout the life of the data. This supports requirements of Title 21 CFR Part 11, Section 11.10, “Revision and change control procedures to maintain an audit trail that documents time-sequenced development and modification of systems documentation.” (21 CFR Part 11 § 11.10)2
5) Implementation to Production
After study-specific configuration is validated, the validator should ensure that the changes were successfully implemented (eg, moved to production). Whether the changes are automatically or manually implemented, testing should verify that configuration matches between environments if applicable (eg, EDC systems vary in how changes are implemented in production). The new or updated software configuration should not be used until confirmation of a successful implementation occurs.
6) Change analysis
A thorough change analysis may be conducted prior to modifying study-specific configuration after the initial implementation. See “Change Control/Maintenance” section under SDLC for more information.
c) Tools/Reports
Tools are often used to make the validation process more efficient. An example of a validation tool would be a report that organizes software configuration settings in a single view for the tester. This may significantly reduce the amount of time to navigate the software to verify the settings. However, software validation tools are subject to the same validation requirements and process as the software itself. Non-validated tools should therefore not be used as a primary source for validation evidence.
12) Use of Real-World Data
Different electronic systems are more readily available to access patient health status; the use of Real World Data is becoming more imminent. But what is Real World Data (RWD) and Real World Evidence (RWE)?
As defined by FDA in “Real World Evidence”, “Real-World Data (RWD) are data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources… Real-World Evidence (RWE) is the clinical evidence regarding the usage and potential benefits or risks of a medical product derived from analysis of RWD.”20
The “Real World Evidence” document indicates that RWD may include data from the following electronic sources:
Electronic health records (EHRs)
Claims and billing activities
Product and disease registries
Patient-generated data including in home-use settings
Data gathered from other sources that can inform on health status, such as mobile devices (Real-World Evidence|FDA)20
It is therefore important for CDM to acknowledge the sources of all data collected, the type of data and how the data will be used to assess the extent of software validation that should be required prior to any analysis. The source and data flow of all systems used in the clinical study should be documented in the data management plan. (Use of Electronic Health Record Data in Clinical Investigations Guidance for Industry (fda.gov) Section V- A)4
a) Certified EHR
For EHR, the “Use of Electronic Health Record Data in Clinical Investigations” guidance indicates that there is no intent for these systems to meet the compliance of 21CFR Part 11 requirements.4 The system, though, should meet specific requirements (45 CFR Part 170)21 and should be certified with The Office of the National Coordinator for Health Information Technology (ONC). (Section V. A of Use of Electronic Health Record Data in Clinical Investigation)4 The certification provides confidence that the data has met minimum requirements for interoperability and verifies that appropriate security measures are in place to protect the privacy of individuals as well as to ensure data integrity of the system. The system should maintain its certification, defined in Subpart D of the 21st Century Cures Act: Interoperability, Information Blocking, and the ONC Health IT Certification Program Final Rule.22 Preference should be given to use of a certified EHR. Confirmation of the system’s certification, carried out by the sponsor, is essential (as recommended per ONC) as it will affirm the validity of the data throughout the lifecycle of the clinical study. Change in certification status may also be a good indicator that system changes may have occurred; in such cases, the system interoperability needs to be re-evaluated and potentially re-verified. When the interoperability of the systems is well defined in the planning phase, a risk-based approach may be considered to evaluate the validation required when a system changes.
b) Non-certified EHR
There are provisions in “Use of Electronic Health Record Data in Clinical Investigations” guidance for non-certified systems usage. These systems may be in place in foreign countries, where the ONC certification is not required. If such a system is utilized in the clinical study, the sponsor’s responsibility should be to determine that appropriate controls are in place to ensure confidentiality, security and data integrity. The sponsor must have adequate processes to still support the interoperability functionality of the systems. The following documentation should be available for review:
“Policies and processes for the use of EHR systems at the clinical investigation site are in place, and there are appropriate security measures employed to protect the study data.
Access to electronic systems is limited to authorized users.
Authors of records are identifiable.
Audit trails are available to track changes to data.
Records are available and retained for FDA inspection for as long as the records are required by applicable regulations.” (Section V.B of the Use of EHR data in Clinical investigations)4
Using either a certified or a non-certified EHR, the sponsor’s responsibility should be to ensure that the interoperability of the systems is functioning as intended and is repeatable, that no data loss occurs, and that data integrity and security are maintained during the exchange of data. Knowing the EHR certification status determines the extent of verification.
c) Other systems
Similar concepts will apply to any other systems (eg, surveys, devices, ePRO) that bring data in the EDC system for RWE use. These systems may be using different platforms (eg, Android, Apple, Fitbit) and interfaces may be implemented to bring data into a structured format. Each system may have a different time frame for the collection of data; and the data may come from different institutions or devices, or may be incomplete. Structured data will avoid having missing data. Analytical tools can be used to identify data issues prior to any analysis. Understanding the data flow and security and reliability of the data will help the assessment of these interfaces’ functionality to confirm the extent of software validation. The FDA and other regulatory agencies will require clear documentation on data source (eg, data flow), data structure (mapping into a data element) as these systems usually have a less structured format (eg, data from randomized clinical trials (RCT)). Adopting a risk-based approach to validation will facilitate the scope of software validation. The key component is being able to show the traceability of the data from collection to analysis to guarantee the source is valid and reliable (2018 Real World Evidence program).23
The extent of interoperability between systems should be assessed as soon as possible (eg, data added, updated, deleted, duplicate records, blinded data, etc.) as this will determine the scope of validation. The CDM role is critical in this assessment: understanding protocol needs, data structure/standards, metadata, and analysis plans so the data can come across appropriately and only inputting the required data points.
The sponsor should ensure that the systems involved have:
Proper documentation by providing the list of all sources used (eg, data management plan) and data flow
Periodic reviews to address any system changes that may affect the interoperability setup and potentially impact data security and data integrity between the systems.
Periodic review of a subset of data integrated in the EDC system is recommended.
Having analytical tools to identify potential data issues (eg, missing data) in place is also recommended (Use of eHR data in Clinical investigation – Section IV. C)4
The validation documentation to assess the interoperability of the system would include similar information as other system(s) (eg, test plan, test summary report, URS, test scripts). Refer to Section 7 of this chapter. The sponsor’s quality system (Use of Electronic Health Record Data in Clinical Investigations Guidance for Industry – Section IV.C)4 (eg, SOPs, SDLC model, change control procedures) should address the interoperability of the EHR and EDC system and the automated electronic transmission of EHR data elements to the EDC system. The main complexity is in the usage of multiple systems to provide RWD for RWE. The CDM should know the data flow to establish if controls are in place from source to data analysis. Each step should be traceable, reliable, secure, and should maintain data integrity. Any changes to these systems may have a greater impact on data integrity at the time of analysis. At the time of writing, when deciding to use RWE to support submission, the recommendation is to involve the regulatory agencies early on to ensure proper design and controls are in place.
13) Recommended SOPs
The following SOPs are recommended in the FDA’s Guidance for Industry: Computerized Systems Used in Clinical Investigations, which states, “The SOPs should include, but are not limited to, the following processes.
System setup/installation (including the description and specific use of software, hardware, and physical environment and the relationship)
System operating manual
Validation and functionality testing
Data collection and handling (including data archiving, audit trails, and risk assessment)
System maintenance (including system decommissioning)
System security measures
Change control
Data backup, recovery, and contingency plans
Alternative recording methods (in the case of system unavailability)
Computer user training
Roles and responsibilities of sponsors, clinical sites and other parties with respect to the use of computerized systems in the clinical trials.”8
ICH E6(R2) section 5.0.1 states that, “During protocol development, the sponsor should identify processes and data that are critical to ensure human subject protection and the reliability of trial results.”3 This implies that organizations should map out the processes involved in study design, start-up, conduct, and closeout and make explicit decisions about which are considered to impact human subject protection and the reliability of trial results. Organizational processes may be partitioned differently leading to different scope and titles for SOPs. Though organizations may differ in how the processes are covered in their SOPs, below is a list of processes commonly considered to impact human subject protection and the reliability of trial results:
Study-specific database design and testing
System validation/documentation (including UAT, risk-based considerations)
Vendor auditing
Interoperability of systems.
14) Literature Review
This revision is based on a systematic review of the peer-reviewed literature indexed for retrieval. The goals of this literature review were to (1) identify published research results and methods of SDLC of the CDM system and (2) identify, evaluate, and summarize the requirements for the conduct of any SDLC system.
The following query was used:
(software[Title/Abstract] or computer[Title/Abstract]) AND (validation[Title/Abstract] or “part 11”[Title/Abstract] or SDLC[Title/Abstract]) AND (“clinical trial”[Title/Abstract] OR “clinical trials” [Title/Abstract] OR “clinical study”[Title/Abstract] OR “clinical studies” [Title/Abstract] OR registry[Title/Abstract] OR registries[Title/Abstract] OR “observational study”[Title/Abstract] OR “interventional study”[Title/Abstract] OR “phase 1 study”[Title/Abstract] OR “phase 2 study”[Title/Abstract] OR “phase 3 study”[Title/Abstract] OR “phase 4 study”[Title/Abstract] OR “phase I study”[Title/Abstract] OR “phase II study”[Title/Abstract] OR “phase III study”[Title/Abstract] OR “phase IV study”[Title/Abstract] OR “first in man”[Title/Abstract] OR “clinical research”[Title/Abstract] OR “device study”[Title/Abstract] OR “interventional trial”[Title/Abstract] OR “phase 1 trial”[Title/Abstract] OR “phase 2 trial”[Title/Abstract] OR “phase 3 trial”[Title/Abstract] OR “phase 4 trial”[Title/Abstract] OR “phase I trial”[Title/Abstract] OR “phase II trial”[Title/Abstract] OR “phase III trial”[Title/Abstract] OR “phase IV trial”[Title/Abstract] OR “randomized clinical trial”[Title/Abstract] OR “clinical research” [Title/Abstract])
The search query was customized for and executed on the following databases: PubMed (82 results); EMBASE (256 results); Science Citation Index/Web of Science (772 results). A total of 1110 works were identified through the searches. The latest search was conducted on February 8, 2022. Search results were consolidated to obtain a list of 1110 distinct articles.
Two reviewers used inclusion criteria to screen all abstracts. Disagreements were adjudicated by the writing group. A total of 36 sources were deemed relevant to SDLC. Of the 36 relevant sources, 6 were identified as informative for the SDLC process. Relevant findings from these three articles have been included in the chapter and graded according to the GCDMP evidence grading criteria as described in Figure 6.
Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) Diagram for Software Development Life Cycle.
15) Revision History
| Publication Date | Comments |
|---|---|
| June 26, 2024 | This Chapter replaces and is a complete revision of the prior chapter titled “Chapter Database Validation, Programming, and Standards.” |
Appendices
Example of User Requirements.
| URS ID | Description | References |
|---|---|---|
| URS1.0 | Security | 21 CFR Part 112 GAMP 56 |
| URS1.1 | Password and user ID must be unique | SOP XX |
| URS1.2 | If the user enters an invalid username/password combination, a warning message must appear | SOP XX |
| URS1.3 | Temporary password expires at first login, forcing user to reset password | SOP XX |
| URS 1.4 | User locked out of the system after X failed attempts | SOP XX |
| URS1.5 | Username must be displayed on the screen while logged in the application | SOP XX |
Example of test script.
| Test Case | Test Script | Step Procedure Topics | Author | Expected Result | Actual Result | Test Run # | Test Run Date | Status |
|---|---|---|---|---|---|---|---|---|
| TC 1 | TS1.1 | Enter username: JohnSmith Password: JohnSmith |
Jane Doe | Warning message appears “Username and password must be unique” | ||||
| TS1.2 | Enter username: JohnSmith Password: j4mth751! |
Jane Doe | Warning message appears “Invalid username and password combination” | |||||
| TS1.3 | Enter username: JohnSmith Password: j4mth751! |
Jane Doe | Password has expired. A new screen appears for password reset. | |||||
| TS1.4 | Enter the following username and password 5 times. Enter username: JohnSmith Password: j4mth752! |
Jane Doe | Warning message appears “Too many unsuccessful logins, the account is locked.” | |||||
| TS1.5 | Enter username: JohnSmith Password: j4mth742! |
Jane Doe | User is able to login and able to see username at the top right corner of the screen |
Traceability Matrix Example at the User Requirement Level.
| Requirement Specification | Test Script | Status | Test Run | Tester Name | Date |
|---|---|---|---|---|---|
| URS 1.1 | TS1.1 | Fail | 1 | John Smith | 01Jan2023 |
| URS 1.1 | TS 1.1 | Pass | 2 | John Smith | 15Jan2023 |
Traceability Matrix Example at the Test Case Level.
| Test Case | Test Script | URS | Test Run # | Test Run Date | Status | Tester Name |
|---|---|---|---|---|---|---|
| Test Case 1 – Security | TS 1.1 | URS 1.1 | 1 | 01Jan2023 | Pass | John Smith |
| TS1.2 | URS 1.2 | 1 | 01Jan2023 | Pass | John Smith | |
| TS1.3 | URS 1.3 | 1 | 01Jan2023 | Pass | John Smith | |
| TS1.4 | URS 1.4 | 1 | 01Jan2023 | Fail | John Smith | |
| TS1.4 | URS 1.4 | 2 | 15Jan2023 | Pass | John Smith |
Disclaimer
Opinions expressed in this article represent the views and opinions of the author and do not express the views or opinions of the author’s employer.
Competing Interests
The authors have no competing interests to declare.
References
1. Advarra 2020 White Paper “Considerations for Using eTools in Research: Part 11 & System Validation” Applied Clinical Trials Online. Published February 28 2020. Accessed 26Jun2023. https://www.appliedclinicaltrialsonline.com/view/considerations-using-etools-research-part-11-system-validation
2. Food and Drug Administration, US Department of Health and Human Services. Electronic Records; Electronic Signatures, 21 CFR §11 (1997). Available from https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm
3. Food and Drug Administration, US Department of Health and Human Services. ICH E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1). March 2018. Available from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e6r2-good-clinical-practice-integrated-addendum-ich-e6r1
4. Food and Drug Administration, US Department of Health and Human Services. Use of Electronic Health Record Data in Clinical Investigations Guidance for Industry. July 2018. Accessed 26Jun2023. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-electronic-health-record-data-clinical-investigations-guidance-industry
5. European Medicines Agency. “Notice to Sponsors on Validation and Qualification of Computerised Systems Used in Clinical Trials”, 7 April 2020 – EMA/INS/GCP/467532/2019. Accessed 26Jun2023. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/notice-sponsors-validation-and-qualification-computerised-systems-used-clinical-trials_en.pdf
6. GAMP® 5 A Risk-based Approach to Compliant GxP Computerized Systems. North Bethesda, MD: International Society for Pharmaceutical Engineering (ISPE). 2008.
7. Food and Drug Administration, US Department of Health and Human Services. General Principles of Software Validation; Final Guidance for Industry and FDA Staff. January 2002. Accessed 26Jun2023. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation
8. Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry: Computerized systems used in clinical investigations. May 2007. Accessed 26Jun2023. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computerized-systems-used-clinical-investigations
9. Food and Drug Administration, US Department of Health and Human Services. Guidance for Industry: Electronic Source Data in Clinical Investigations. September 2013. Accessed August 8, 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/electronic-source-data-clinical-investigations
10. Von Culin R. New Approach to System Validation. Appl Clin Trials Online. 2011; 20(2): 32–37. Accessed 26Jun2023. https://www.appliedclinicaltrialsonline.com/view/new-approach-system-validation
11. Nidagundi P, Novickis L. Introduction to Lean Canvas Transformation Models and Metrics in Software Testing. Applied Computer Systems. Published Online. 2016; 19(1): 30–36. DOI: http://doi.org/10.1515/acss-2016-0004
12. Public Welfare, US Department of Health and Human Services. Security Standards for the Protection of Electronic Protected Health Information, 45 CFR § 164.308; October 2007 (Up to date as of 7/14/2023). Accessed May 2, 2023. https://www.ecfr.gov/current/title-45/section-164.308
13. IEEE Standard for Software and System Test Documentation. IEEE Std 829-2008. Published online July 1, 2008; 1–150. DOI: http://doi.org/10.1109/IEEESTD.2008.4578383
14. Grassi PA, Fenton JL, Newton EM, et al. Digital identity guidelines: authentication and lifecycle management. U.S. Department of Commerce, National Institute of Standards and Technology. Published online June 22, 2017. Accessed 26Jun2023 DOI: http://doi.org/10.6028/NIST.SP.800-63b
15. International Organization for Standardization, Security and Resilience. Business continuity management systems. Guidance on the use of ISO 22301. Published online February 28, 2020. Accessed 26 Jun 2023. DOI: http://doi.org/10.3403/30379713
16. International Council for Harmonisation (ICH), Quality Risk Management Q9(R1), November 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use-ich-guideline-q9-quality-risk-management-step-5-first-version_en.pdf
17. Peterson D. A Sponsor’s Role: 21 CFR Part 11. Data Basics. 2016; 22(3): 4–5. Accessed 26 Jun 2023. https://www.prometrika.com/pdf/db2016Fall.pdf
18. European Commission Health and Consumers Directorate-general. EudraLex; The Rules Governing Medicinal Products in the European Union Volume 4: Annex 11: Computerised Systems (2011). Annex 11 Final 0910 (europa.eu). https://ec.europa.eu/health/documents/eudralex/vol-4_ga
19. US Department of Health & Human Services. Summary of the HIPAA Security Rule. HHS.gov. Published October 19, 2022. Accessed 26Jun2023. https://www.hhs.gov/hipaa/for-professionals/security/laws-regulations/index.html
20. US Food and Drug Administration, Office of the Commissioner. Real-World Evidence, 2019. Accessed 26Jun2023. https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence
21. US Department of Health and Human Services. Health Information Technology Standards, Implementation Specifications, and Certification Criteria and Certification Programs for Health Information Technology, 45 CRF Part 170. (Last amended 7/13/2023). Accessed 26Jun2023. https://www.ecfr.gov/current/title-45/subtitle-A/subchapter-D
22. US Department of Health and Human Services. 21st Century Cures Act: Interoperability, Information Blocking, and the ONC Health IT Certification Program, 85 FR 25642, (Final Rule effective June 30, 2020). Accessed 26Jun2023. https://www.federalregister.gov/documents/2020/05/01/2020-07419/21st-century-cures-act-interoperability-information-blocking-and-the-onc-health-it-certification
23. US Food and Drug Administration. Framework for FDA’s Real-World Evidence Program. December 2018. https://www.fda.gov/media/120060/download