

Background
The World Health Organization (WHO) declared the outbreak of Coronavirus Disease 2019 (COVID-19) a pandemic on March 11, 2020.1 The U.S. President declared a national emergency in response to COVID-19 on March 13, 2020.2 The pandemic limited the ability of regulatory agencies to conduct on-site good clinical practice (GCP) inspections due to lockdowns and other travel restrictions, as well as the need to protect inspectors and the site staff. Regulatory agencies around the world swiftly responded to the pandemic, adapting to the public health crisis and conducting inspection activities to ensure continued regulatory oversight of clinical trial participant safety, data integrity, and GCP compliance.3,4,5,6,7,8,9,10
As part of the Bioresearch Monitoring (BIMO) program at the U.S. Food and Drug Administration (FDA), GCP inspections of clinical investigators (CIs)11 and sponsors and contract research organizations (CROs)12 are conducted to assess the quality and integrity of data submitted to the FDA, as well as compliance with the regulations that govern the conduct of clinical trials. GCP inspections ensure the reliability of safety and efficacy data of clinical trials submitted to the FDA in support of marketing applications.
At the onset of the COVID-19 public health emergency, the FDA limited on-site GCP inspections to those applications considered mission critical, based on factors such as whether the product had received breakthrough therapy designation or would be used for a serious disease with no current available treatments.13,14 For these applications, the feasibility of on-site inspections was assessed based on factors such as lockdowns, other travel restrictions, and COVID-19 infection rates provided by the COVID-19 Advisory Rating System.15
When on-site GCP inspections were not feasible, the FDA used innovative alternative tools to evaluate data integrity of clinical trials to help inform regulatory decisions for New Drug Applications (NDAs) and Biologics License Applications (BLAs). These alternative tools included remote regulatory assessments (RRAs), a remote evaluation of an FDA-regulated establishment and/or its records for data integrity and compliance with relevant FDA regulations, as well as review of foreign regulatory counterpart inspection reports.16,17,18
In this paper, we present our experience in using alternative tools to evaluate data integrity for pivotal Phase II/III clinical trials supporting marketing applications during the COVID-19 public health emergency in the Office of Scientific Investigations (OSI) in the Center for Drug Evaluation and Research (CDER). In addition, we analyze the impact of the pandemic on on-site GCP inspections and discuss the contribution of alternative tools to GCP activities and the implications for the future.
Methods
The main data sources for this study included a CDER internal database, Compliance Program Information System (COMPLIS), Enterprise Content Management System (ECMS—document repository), and the Document Archiving, Reporting, & Regulatory Tracking System (DARRTS). The information collected included marketing applications, product types, and inspection activity specifics, such as inspection type, site location, and mission priority, as well as whether on-site inspections or remote evaluations using alternative tools were conducted.
The Office of Regulatory Affairs conducted the first RRA for OSI on April 13, 2020, a remote evaluation of a U.S. CI in support of a mission critical BLA for an oncology product. We identified instances when alternative tools were used in lieu of on-site GCP inspections from April 13, 2020, through September 30, 2021. We then obtained the memoranda of these evaluations in ECMS and conducted a review to ensure the accuracy of the number and type of alternative tools used during the study period.
We reviewed, described, and characterized the alternative tools used in lieu of on-site GCP inspections in FY2020 and FY2021. In addition, we reviewed RRA memoranda and examined their scope and content, as well as obtained the total number of on-site GCP inspections from FY2015 to FY2021 from COMPLIS. Fiscal year is defined here as October 1 to September 30. We compared the total number of on-site GCP inspections in FY2020 and FY2021 to the yearly average for the five years preceding the pandemic (FY2015 through FY2019) and calculated the contribution of the use of alternative tools to the GCP activities in FY2020 and FY2021.
Results
In FY2020, 39 GCP evaluations were conducted using alternative tools to support 13 NDAs and 7 BLAs from 13 clinical divisions in the Office of New Drugs (OND). The top two therapeutic areas for which these evaluations were conducted were oncology and anti-viral, with five and three applications, respectively. In FY2021, 38 GCP evaluations were conducted using alternative tools to support 16 NDAs and 5 BLAs from 13 clinical divisions in OND. The two top therapeutic areas were oncology and rare diseases/medical genetics, with three applications each.
The FDA used alternative tools to evaluate the reliability and integrity of clinical trial data submitted in support of marketing applications when on-site GCP inspections could not be completed. The alternative tools included 1) remote evaluations conducted from FDA locations, 2) remote evaluations of non-U.S. sites conducted from U.S. sponsor or agent locations, 3) reviews of study records obtained from sponsors through the FDA’s information request, and 4) review of inspection reports shared by a foreign regulatory counterpart. The type and number of alternative tools and the relevant characteristics of the GCP evaluations conducted using these tools are shown in Table 1 and Table 2, respectively.
Alternative Tools Used for GCP Evaluations in Fiscal Years 2020 and 2021.
| Type of Alternative Tool | FY2020 N (%) |
FY2021 N (%) |
Total N (%) |
|---|---|---|---|
| Remote Evaluations from FDA Locations | 28 (72%) | 25 (66%) | 53 (69%) |
| Remote Evaluations of Non-U.S. Sites from U.S. Sponsor or Agent Locations | 10 (26%) | 6 (16%) | 16 (21%) |
| Review of Study Records Obtained from Sponsors Through FDA’s Information Request | 1 (2%) | 2 (5%) | 3 (4%) |
| Review of Inspection Reports Shared by European Medicines Agency | 0 | 5 (13%) | 5 (6%) |
Characteristics of GCP Evaluations Conducted Using Alternative Tools in Fiscal Years 2020 and 2021.
| Characteristics of Evaluations | FY2020 N = 39 |
FY2021 N = 38 |
Total N = 77 |
|---|---|---|---|
| New Drug Application (NDA) | 23 (59%) | 32 (84%) | 55 (71%) |
| Biologics License Application (BLA) | 16 (41%) | 6 (16%) | 22 (29%) |
| Original Submission | 33 (85%) | 29 (76%) | 62 (81%) |
| Supplemental Submission | 6 (15%) | 9 (24%) | 15 (19%) |
| Evaluations of Clinical Investigators | 33 (85%) | 28 (74%) | 61 (79%) |
| Evaluations of Sponsors/CROs | 6 (15%) | 10 (26%) | 16 (21%) |
| Sites Located Outside United States | 33 (85%) | 27 (71%) | 60 (78%) |
| Sites Located in the United States | 6 (15%) | 11 (29%) | 17 (22%) |
| Mission Critical | 20 (51%) | 27 (71%) | 47 (61%) |
| Non-Mission Critical | 19 (49%) | 11 (29%) | 30 (39%) |
CRO = contract research organization.
Each alternative tool used during the COVID-19 public health emergency is described in detail below.
1) Remote Evaluations from FDA Locations
The FDA conducted remote evaluations from FDA locations of CIs, sponsors, and CROs, located at both US and non-US sites, in accordance with relevant BIMO compliance programs. Remote evaluations were voluntary and were conducted using technologies such as teleconference, video conference, data sharing via an on-line platform, and read-only remote access to documents such as individual subject records and the trial master file. The remote evaluations described in this paper were voluntary. An establishment could decline to participate or withdraw participation during the voluntary remote evaluation, in which case the Agency would consider other tools for evaluating compliance with FDA requirements.18 This was the most used alternative tool in FY2020 and FY2021, with 28 remote evaluations from FDA locations conducted in FY2020 and 25 conducted in FY2021, accounting for 72% and 66% of the total GCP evaluations conducted using alternative tools, respectively (Table 1).
A total of 40 remote evaluations were conducted from FDA locations of CI sites in FY2020 and FY2021. Review of the RRA memoranda found that FDA inspectors were able to review source data to demonstrate participants met the eligibility criteria at all CI sites. FDA inspectors were also able to access the adverse event source data to verify safety data reporting for all but one CI. They were also able to verify the primary efficacy endpoint data for the respective marketing applications for 38 of the 40 CIs. For one CI, the primary efficacy endpoint data were not assessed due to time constraints and staffing issues. At this site, the data were recorded in a logbook that contained results from other patients who didn’t participate in the study, making identification of study subjects cumbersome. For the other CI site, the primary efficacy endpoint data were centrally reviewed and adjudicated and not available at the site.
A total of nine remote evaluations were conducted of sponsors and four of CROs in FY2020 and FY2021. The review of the sponsor RRA memoranda found that during all evaluations FDA inspectors were able to review the electronic trial master file, transfers of obligations to contractors, data management, monitoring, quality assurance, and study drug handling and accountability. The review of the CRO RRA memoranda found that the FDA inspectors were able to review the tasks and responsibilities for which the CROs were contracted. The availability of electronic systems at the sponsor/CRO sites facilitated the review of data and documents for the evaluations.
2) Remote Evaluations of non-U.S. sites from U.S. sponsor or agent locations
The FDA conducted remote evaluations of non-US CIs from US sponsor locations as well as an evaluation of a non-US sponsor from its US agent location (Table 1).
In FY2020, the FDA conducted 10 remote evaluations of non-US CIs located in the Democratic Republic of the Congo (8), Argentina (1), and Columbia (1) from their US sponsor locations due to travel restrictions to Argentina during the COVID-19 pandemic and FDA restrictions on conducting inspections in Colombia and the Congo. The FDA worked with the sponsors to obtain certified copies of the original paper medical records and other paper source documents for all subjects screened and enrolled at these sites, as well as certified copies of any electronic records, including electronic case report forms. The sponsors then uploaded the records to a cloud service provider and made the files available to FDA inspectors through a secure file sharing system. During these evaluations, CIs were available to participate in meetings via telephone to answer study-specific questions. Sponsor representatives and translators (provided by the sponsors) were present to assist with the evaluations.
In FY2021, the FDA conducted six remote evaluations of non-US CIs from US sponsor or agent locations, including five for non-US CIs from their US sponsor locations and one for a non-US sponsor from its US agent’s location (Table 1). All six non-US sites were in the EU, where the General Data Protection Regulations (GDPR) limited the transmission of identifiable health data outside the EU. For the evaluations of the five CIs, source document review was conducted by the CI staff viewing the source documents and then screen sharing with the FDA inspectors at the US sponsor locations. The evaluation of the non-US sponsor was conducted remotely via teleconferencing and screen sharing from its US agent’s location.
3) Remote review of study records obtained through the FDA’s Information Request
The FDA conducted remote reviews of study records obtained through an FDA information request for three non-US CIs located in Poland, Canada, and South Korea when on-site inspections were not feasible from October 2020 to March 2021 due to the pandemic and conducting a comprehensive remote evaluation was limited due to reasons such as staffing issues or institutional restrictions (Table 1). In these instances, the FDA made voluntary information requests to the sponsor. FDA inspectors then reviewed the study records obtained from the sponsors, which were limited to electronic case report forms, monitoring reports, and lab reports.
This remote review method was different from the two types of RRAs described above because the records reviewed were limited to what the sponsor could provide, and there were no interactions with CIs via teleconference or otherwise.
4) Review of inspection reports shared by a foreign regulatory counterpart
For one marketing application, the FDA reviewed inspection reports shared by the European Medicines Agency (EMA) in 2021. In this case, marketing applications, containing the same clinical trial data, were under review by both the FDA and the EMA. The EMA had already conducted five on-site GCP inspections of three CIs and two sponsors for this application and provided the individual inspection reports and the integrated inspection report to the FDA. The FDA reviewed and assessed the EMA’s findings to determine the potential impact on data integrity and subject safety (Table 1).
Total GCP on-site inspections in FY2015 through FY2021
The total number of on-site GCP inspections of CIs, sponsors, and CROs for marketing applications conducted from FY2015 through FY2021 are depicted in Figure 1. On average, there were 481 GCP inspections conducted yearly during the five years preceding the pandemic (FY2015 through FY2019). The total number of on-site GCP inspections conducted in FY2020 was 370 and in FY2021 was 451, which was a 23% and 6% decrease, respectively, compared to the pre-pandemic yearly average of 481.
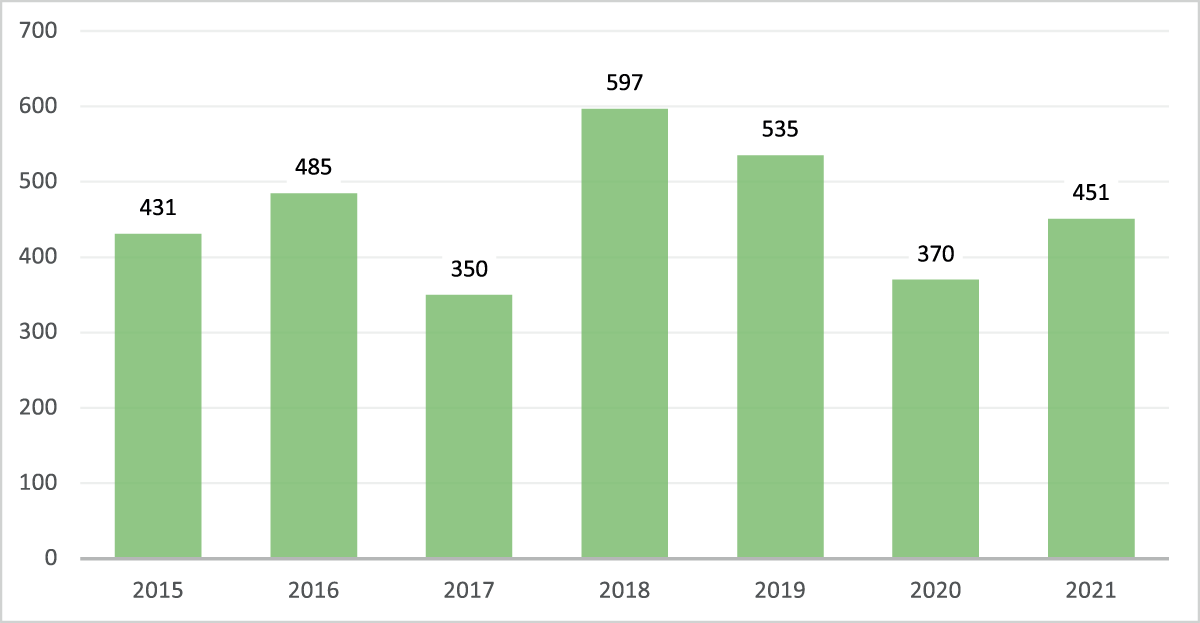
The Total Number of On-site Good Clinical Practice (GCP) Inspections for Clinical Investigators, Sponsors and Contract Research Organizations for CDER Marketing Applications in Fiscal Years 2015–2021.
This graph demonstrates the pandemic impact on on-site GCP inspections. On average, there were 481 on-site GCP inspections conducted yearly from FY2015 to FY2019. The total number of on-site GCP inspections conducted in FY2020 and FY2021 was 370 and 451, representing a 23% and 6% decrease, respectively, compared to the yearly average of 481 for FY2015 through FY2019.
Total GCP activities in FY2020 and FY2021
Total GCP activities include on-site inspections as well as GCP evaluations conducted using alternative tools. In FY2020, the total number of GCP activities was 409, including 370 (90%) on-site inspections and 39 (10%) GCP evaluations using alternative tools. In FY2021, the total number of GCP activities was 489, including 451 (92%) on-site inspections and 38 (8%) GCP evaluations using alternative tools. The alternative tools used in FY2021 included the review of five inspection reports (three CIs and two sponsors) shared by the EMA (Figure 2).
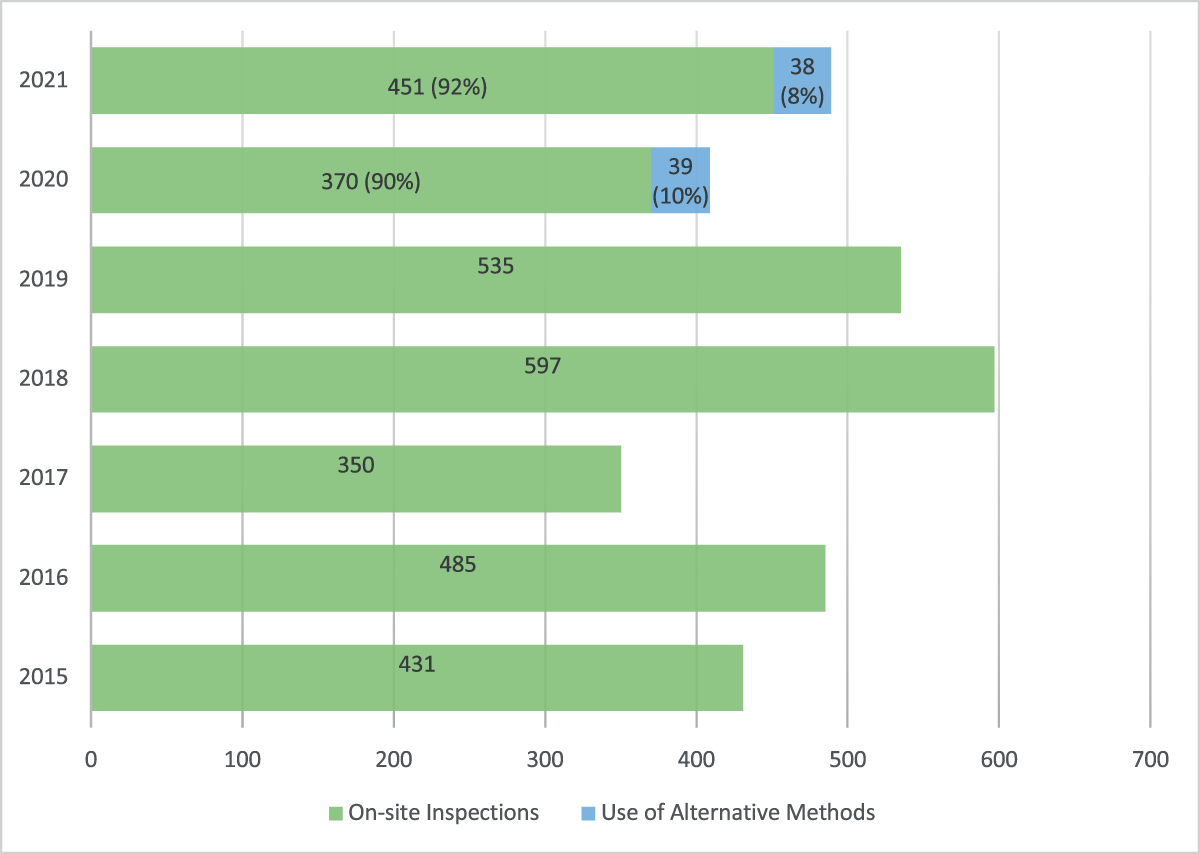
The Total Number of Good Clinical Practice (GCP) Activities Conducted for Clinical Investigators, Sponsors and Contract Research Organizations for Marketing Applications in Fiscal Year 2015–2021.
GCP activities included on-site GCP inspections and GCP evaluations using alternative methods. Alternative methods contributed to 10% and 8% to total GCP activities in FY2020 and FY2021, respectively.
Discussion
Alternative tools were utilized for 39 GCP evaluations in FY2020 and 38 in FY2021, and the majority were RRAs of non-US CIs supporting mission critical original NDAs. The use of alternative tools also played an important role in data integrity assessments for non-mission critical applications. In FY2020, almost half of the GCP evaluations conducted using alternative tools were for applications deemed non-mission critical. The most commonly used alternative tool was remote evaluations from FDA locations. The review of these RRA memoranda of the evaluations from FDA locations for CIs found that this type of RRA appeared to be useful to assess GCP documentation related to study eligibility, safety reporting, and primary efficacy endpoints. In addition, review of the sponsor/CRO RRA memoranda suggests that the remote review of electronic records is useful in assessing their responsibilities and conduct of clinical trials.
COVID-19 pandemic related travel restrictions impacted the total number of on-site GCP inspections conducted in FY2020 (370) and FY2021 (451), a 23% and 6% decrease, respectively, compared to the yearly average of 481 on-site GCP inspections in the five years preceding the pandemic. The FDA transitioned to standard operating levels for domestic inspections in July 2021,19 which was likely facilitated by the increased availability of COVID-19 vaccines, diagnostics, and therapeutics. This was evidenced by the increase in the total number of on-site GCP inspections from 370 in FY2020 to 451 in FY2021 (Figure 1).
FDA used alternative tools to accomplish 10% and 8% of the total GCP activities in FY2020 and FY2021, respectively (Figure 2). The GCP evaluations conducted using alternative tools were able to inform the Agency’s regulatory decisions for 13 NDAs and 7 BLAs in FY2020 and 16 NDAs and 5 BLAs in FY2021. Therefore, the use of alternative tools in lieu of on-site GCP inspections played a significant role in ensuring the reliability of data submitted in marketing applications.
The decision to use alternative tools was made on a case-by-case basis and depended on the inspection types, sites, and the laws of local jurisdictions. Although the use of RRA was important to ensure continuity of GCP oversight during the COVID-19 pandemic, it has several limitations: 1) feasibility depended on technology issues such as the quality of the internet connection; 2) the need for CI site staff to redact source records and upload them to an online platform was time consuming and may have interfered with subject care responsibilities, particularly when sites were already experiencing stressed resources due to the COVID-19 pandemic; 3) time zone differences for non-US remote evaluations; 4) translation issues; 5) unclear views of documents when using screen sharing; 6) data protection regulations limiting transmission of identifiable health data outside the EU; 7) inability to walk through the pharmacy and the study drug storage areas. Nevertheless, the use of RRAs enabled the Agency to verify efficacy and safety data to inform regulatory decisions for marketing applications when on-site GCP inspections were not possible. It also helped protect FDA inspectors and the site staff from COVID-19 by minimizing travel and in-person interactions.
Conclusions
The innovative alternative tools used by the FDA to evaluate data reliability and integrity, GCP compliance, and subject safety during the COVID-19 public health emergency have been critical in informing the agency’s regulatory decisions for marketing applications while mitigating the spread of COVID-19. Our study shows that the remote evaluation of clinical trials from FDA locations was useful in evaluating CI and sponsor/CRO data integrity, subject safety, and clinical trial conduct. Looking forward, alternative tools can be complementary to on-site GCP inspections, particularly when travel limitations exist. In addition, these tools could be considered to expand the breadth of the inspection coverage.
Acknowledgements
We thank Yolanda Patague for recording GCP inspection activity data; Amir Tahami, M.B.A., for assisting in obtaining the total number of GCP on-site inspection data for the study. We also thank Stefanie Krause, J.D.; Nancy Hayes, J.D.; and Cheryl Grandinetti, Pharm.D. for their review of the draft manuscript; all FDA inspectors from the Office of Regulatory Affairs (Office of Bioresearch Monitoring Operations) for the GCP inspections they conducted; and all the GCP Assessment Branch staff for their collaboration with internal and external stakeholders for the GCP evaluation work in support of marketing applications.
Funding Information
No financial support was received for the research, authorship, and/or publication of this article.
Competing Interests
The authors declare no conflicts of interest. The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the agency with which each author is affiliated..
References
1. World Health Organization. Coronavirus disease 2019 (COVID-19) Situation Report 51. https://www.who.int/publications/m/item/situation-report---51 Accessed June 30, 2020.
2. The White House. Notice on the Continuation of the National Emergency Concerning the Coronavirus Disease 2019 (COVID-19) Pandemic. https://www.whitehouse.gov/briefing-room/presidential-actions/2022/02/18/notice-on-the-continuation-of-the-national-emergency-concerning-the-coronavirus-disease-2019-covid-19-pandemic-2/. Accessed June 20, 2022.
3. Bolislis WR, de Lucia ML, Dolz F, et al. Regulatory Agilities in the Time of COVID-19: Overview, Trends, and Opportunities. Clin Ther. 2021 Jan; 43(1): 124–139. Accessed June 20, 2022. DOI: http://doi.org/10.1016/j.clinthera.2020.11.015
4. Mofid S, Bolislis WR, Brading C, et al. The Utility of Remote Inspections During the COVID-19 Health Emergency and in the Post pandemic Setting. Clin Ther. 2021 Dec; 43(12): 2046–2063. DOI: http://doi.org/10.1016/j.clinthera.2021.10.003
5. European Medicine Agency (EMA). Guidance on remote GCP inspections during the COVID19 pandemic. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/guidance-remote-gcp-inspections-during-covid-19-pandemic_en.pdf. Accessed June 20, 2022.
6. Medicines and Healthcare products Regulatory Agency (MHRA). Guidance for industry on MHRA’s expectations for return to UK on-site inspection. 2020. https://www.gov.uk/guidance/guidance-for-industry-on-mhras-expectations-for-return-to-uk-on-site-inspections. Accessed June 20, 2022.
7. Churchward D. MHRA Good Practice (GxP) inspections during the COVID19 outbreak. 2020. https://mhrainspectorate.blog.gov.uk/2020/03/23/mhra-good-practice-gxp-inspections-during-the-covid19-outbreak/. Accessed June 20, 2022.
8. Pharmaceuticals and Medical Devices Agency (PMDA), Japan. Procedure for Remote Inspection as a part of compliance inspection on drugs and regenerative medical products. 2020. https://www.pmda.go.jp/files/000238733.pdf. Accessed June 20, 2022.
9. Walker P. Inspectors grounded – a year of innovation. 2021. https://mhrainspectorate.blog.gov.uk/2021/03/26/inspectors-grounded-a-year-of-innovation/. Accessed June 20, 2022.
10. International Coalition of Medicines Regulatory Authorities (ICMRA) COVID 19 Working Group. Reflections on the regulatory experience of remote approaches to GCP and GMP regulatory oversight during the COVID-19 Pandemic. 2021 https://www.icmra.info/drupal/sites/default/files/2021-12/remote_inspections_reflection_paper.pdf. Accessed June 20, 2022.
11. FDA. Food and Drug Administration Compliance Program. Bioresearch Monitoring. Subject: Clinical Investigators and Sponsor-Investigators. 07/22/2020. https://www.fda.gov/media/75927/download. Accessed June 20, 2022.
12. FDA. Food and Drug Administration Compliance Program. Bioresearch Monitoring. Subject: Sponsors and Contract Research Organizations. September 15, 2021. https://www.fda.gov/media/75916/download. Accessed June 20, 2022.
13. FDA. U.S. FDA’s Ongoing Use of Inspectional Tools for Ensuring Access to Safe, Quality Food and Medical Products During the COVID-19 Pandemic. 2021. https://www.fda.gov/news-events/fda-voices/fdas-ongoing-use-inspectional-tools-ensuring-access-safe-quality-food-and-medical-products-during. Accessed June 20, 2022.
14. FDA. Resiliency Roadmap for FDA Inspectional Oversight. May 2021. https://www.fda.gov/media/148197/download. Accessed June 20, 2022.
15. FDA. Coronavirus (COVID-19) Update: FDA prepares for resumption of domestic inspections with new risk assessment system. 2020. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-prepares-resumption-domestic-inspections-new-risk-assessment-system. Accessed June 20, 2022.
16. FDA. Manufacturing, Supply Chain, and Drug and Biological Product Inspections During COVID-19 Public Health Emergency. Questions and Answers. Guidance for Industry. August 2020. Updated on May 17, 2021. https://www.fda.gov/media/141312/download. Accessed June 20, 2022.
17. FDA. Remote Interactive Evaluations of Drug Manufacturing and Bioresearch Monitoring Facilities During the COVID-19 Public Health Emergency Guidance for Industry. 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/remote-interactive-evaluations-drug-manufacturing-and-bioresearch-monitoring-facilities-during-covid. Accessed June 20, 2022.
18. FDA. Conducting Remote Regulatory Assessments. Questions and Answers. Draft Guidance for Industry. July 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/conducting-remote-regulatory-assessments-questions-and-answers. Accessed August 9, 2022.
19. FDA. An Update to the Resiliency Roadmap for FDA Inspectional Oversight. November 2021. https://www.fda.gov/media/154293/download?utm_medium=email&utm_source=govdelivery. Accessed June 20, 2022.